
1. INTRODUCTION
Prostate cancer (PCa) is a hereditary disease, and its prevalence rate is more common in men aged 70 years and above. Many obstacles have been placed in the way of the search for an etiological cause of PCa by the heterogeneity of the gland itself [1]. PCa stands out as one of the most prevalent malignancies and the second most common cancer among men followed by lung cancer in terms of fatality [2]. Globally, medicinal plants are now widely used, and in current years, they become crucial in the treatment of ailments [3]. The side effects of cancer medications have been traditionally minimized using different plant extracts due to their cost effectiveness, efficacy, easy accessibility, and preparation. Currently, several anti-cancer drugs have been developed from plants such as paclitaxel and taxol from Taxus brevifolia, vincristine and vinblastine from Catharanthus roseus and docetaxel (Taxotere) from Taxus baccata [4].
Flax is a multipurpose crop in the Linaceae family. The scientific name of flax “usitatissimum” originated from Latin which means “the most useful” one [5]. The health benefits of flax/flaxseed have drawn the researcher’s attention (Fig. 1), which include cancer treatment and prevention [6]. The seeds are abundant in lignans, dietary fiber, and essential fatty acids, and have been demonstrated to have anti-cancerous effects [7]. Secoisolariciresinol glycoside (SDG) is the most prevalent lignan of flax, when consumed, it will metabolize by the gut microbiota and produces two mammalian lignans, Enterodiol and Enterolactone [8]. Other lignans include isolariciresinol, anhydrosecoisolariciresinol, matairesinol pinoresinol, and pinoresinol diglucoside [9]. Several experimental studies in the past have documented the importance of flax/flaxseeds and their phytoconstituents in retarding the progression of different cancers [10]. Previous studies have shown that the flaxseeds in the diet reduce the risk of PCa by significantly decreasing the proliferation effect and the levels of prostate-specific antigen in Wistar rats [11]. According to Johnsson [12], the flax meal diet supplemented with seeds of flax showed anti-cancerous activity on colon cancer in rats. It has also been reported that flax extract containing enterolignans significantly reduced the risk of breast cancer in subjects [12]. Additionally, research findings by Saggar et al. [13] and Viveky et al. [14] also revealed that flaxseed consumption led to a reduction in tumor growth rate in ovarian cancer subjects thereby exhibiting an anti-proliferative effect. Many lignans found in flaxseed suppress the important regulatory proteins and alter apoptotic pathways to specifically target cancer cells [15]. The class of cysteine proteases known as caspases (cysteine aspartyl-specific proteases) cleaves target proteins to cause apoptosis. [16]. When apoptotic control is lost, cancer cells are able to live longer and accumulate, this can lead to angiogenesis, an increase in tumor growth as well as cell proliferation and differentiation derangement [17]. Despite the fact that dietary lignans upregulate the estrogen receptor-mediated pathway and are, therefore, advised against consumption by patients with PCa, some lignans, such as SDG or trans-(-)-Hinokinin, have been shown to have antitumor effects on breast cancer cells through the inhibition of Akt, Cyclin D1, and CDK protein targets [18]. Progress through the four important stages of the cell cycle—G0/G1, S, G2, and M—is necessary for proliferation. These phases are controlled by a number of cyclin-dependent kinases, which function in complex with their cyclin partners to maintain genetic material duplication and cell division. The cell cycle derangement caused by abnormal expression of key regulatory proteins in the cell cycle is closely linked to the development of tumors [19]. Dibenzylbutyrolactol, a major lignan of flax showed strong anti-proliferative activity by arresting the G2/M checkpoint and enhancing the protein expression of the G2/M phase in human A549 and CL1-5 cells [20].
 | Figure 1. Diagrammatic representation of medicinal uses of flaxseed lignans. [Click here to view] |
Even though early research on the anti-proliferative properties of lignans derived from flaxseeds has attracted attention and is somewhat encouraging, further investigation, especially in clinical settings, is necessary to support the viability of Linum usitatissimum as a PCa therapy. In this study, we conducted molecular docking simulation to confirm the potential interactions between the identified phytoconstituents of flax microgreens and PCa target proteins as well as to understand the basic mechanisms in which these compounds fit into the binding pocket of specific receptor molecules. The knowledge gathered from this study will not only help to clarify the medicinal potential of flax microgreens (L. usitatissimum), but also lay the foundation for future research on the physiological mechanisms of action of lignans in the treatment of PCa.
2. MATERIAL AND METHODS
2.1. Plant Material and Growth Conditions
The trays were filled with coco peat and the flaxseeds were sprinkled evenly and thickly over it. The flaxseeds were misted gently with a plant sprayer to avoid over watering and preventing the seed from being displaced. The trays were placed in a warm and well-lit area (Hi-Tech polyhouse 26°C to 30°C during daytime and 15°C to 18°C at night) to avoid direct sunlight as it can dry out the coco peat. The coco peat’s moisture level was checked daily and misted carefully to avoid overwatering. The microgreens were harvested by cutting them just above the surface of the coco peat with clean scissors.
2.2. Collection of Plant Material
Fax microgreens were collected from Hi-Tech Polyhouse (equipped with a fan pad system for cooling, thermo-regulation, and misting facility for maintaining humidity inside the chamber), Lovely Professional University (LPU) Phagwara, Punjab-India. The collected microgreens were washed to make them free from cocopeat and shade dried at room temperature.
2.3. Extraction by Cold maceration Technique
The powdered sample of flax microgreens was extracted using methanol. About 50 g of plant samples were extracted in 250 ml of solvent by cold maceration method. The powdered plant materials were kept in a conical flask in contact with the solvent in a stopped container for 24 hours with frequent agitation and then filtered using Whatmann filter paper no. 1. Using a rotary evaporator, the extract was concentrated at 40°C under reduced pressure to produce a viscous semi-solid material [21].
2.4. Preliminary Phytochemical Screening
The qualitative phytochemical screening of methanolic extract of flax microgreens was determined as previously described by Velavan [22] and Rajasree et al. [23].
2.5. Gas Chromatography–Mass Spectrometry Analysis
The Shimadzu (GCMS-TQ8040 NX) Gas Chromatograph was used for the GC-MS study. It was connected to a Perkin Elmer Turbomass 5.1 mass detector Turbo mass gold with an Elite 1 (100% Dimethyl poly siloxane) capillary column measuring 30 m × 0.25 mm ID × 0.25 μm. The temperature of the instrument was initially set to 50°C, and it remained there for 3 minutes. The oven temperature was increased at the rate of 10°C/minute, rosed up to 300°C and maintained for 8 minutes. Injection port temperature was ensured at 250°C and helium flow rate at 1.02 ml/minute. The ionization voltage was 70 eV. The split mode of injection for the samples was 10:1. The range of the mass spectral scan was 34600 (m/z). It kept the interface temperature at 310°C and the ion source temperature at 240°C. The MS start time was 4 minutes, and the end time was 37 minutes with a solvent cut time of 4 minutes. The contents of phytochemicals present in the test sample were identified based on the comparison of peak area, peak height, retention time (minutes), and mass spectral patterns with those spectral databases of authentic compounds stored in the National Institute of Standards and Technology (NIST) library [24].
2.6. Protein Modeling Studies of Delta-Like Ligand 3 (DLL3)
2.6.1. Delta-like ligand 3 sequence recovery
The Delta-Like Ligand 3 (DLL3) sequence of humans was obtained in FASTA format from the UniprotKB database with sequence identity (ID: Q9NYJ7) [25]. The ExPASy ProtParam server was used to compute the physicochemical properties of protein sequence which include molecular weight (MW), isoelectric point (pI), instability index (II) [26], total number of amino acids residues, grand average hydropathy (GRAVY) [27], extinction coefficient [28], and aliphatic index (AI) [29–31]. The InterPro server and Conserved Domain Database (CDD) of the NCBI were used to confirm the protein domains and functional sites after they were identified using the Prosite database [32,33]. PSSpred and SOPMA online tools were used to study and analyze the structural features of the DLL3 [34,35]. The protein conformation was selected viz. alpha-helix, beta-sheet, coil and turn, window width, and similarity threshold were maintained at 17 and 8, respectively, whereas 50 output with used for SOPMA. The transmembrane regions were identified using TMHMM 2.0 [36]. Important features like solvent accessibility, amino acid arrangement, secondary structure, and their composition were assessed using the PredictProtein server. The buried hydrophobic and exposed hydrophilic regions reflected the solvent accessibility [37].
2.6.2. Protein modeling
The SWISS-MODEL online software was used to generate 3D structure of protein target DLL3 [38]. Comparative protein modeling is carried out by the SWISS-MODEL using fragment-based assembly and local similarity search. Protein threading, homology modeling, and ab-initio are the protein modeling techniques used by the SWISS-MODEL. After superimposing the two created models, a standard deviation and distance plot were produced. Ramachandran plot analysis was used to study the stereochemical characteristics of the modeled protein [39]. The overall structural quality of the modelled DLL3 was obtained using SAVES v6.0 server with PROCHECK, VERIFY 3D, and PROVE [40–42]. The UCSF Chimera 1.13.1 was used to generate publication-quality images and structural analysis [43].
2.7. In silico Analysis and Biomolecular Interactions
A molecular docking simulation was used to study the protein-ligand interaction using PyRx tool which is a virtual screening tool that employs Vina as well as Autodock 4.2 [44,45]. The phytochemicals and three FDA-approved drugs in .sdf format were obtained from the PubChem database (www.pubchem.com) and converted into .pdbqt format before running docking [46,47]. The 3D structure of target proteins was downloaded from the protein databank (http://www.rscb.org/pdb) and using grid box analysis, the active site was manually predicted [48]. Protein configurations were improved by removing extraneous water molecules and the only area essential for binding with ligands was kept. The optimal geometries for docking scenarios were obtained by ligand optimization. The docking begins as soon as the ligands and proteins are ready. In this stage, the ligands were bound into the active side of the protein and the binding affinity was measured. Through PyRx AutoDock Vina simulations, the binding affinity strengths and patterns were attained, and protein-ligand interaction was understood and identified. After the docking search was completed, the Protein-Ligand interaction profiler (PLIP), PyMOL, and LigPlot software were used to study protein-ligand interactions in the pdb format preparations [49–51]. The ligand’s binding strength was determined using a negative score (kcal/mol) [52]:
Ki = e–?DG/RT
where ?G = Gibbs free energy; R = (1.985 × 10-1 kcal/mol/K); T = (298.15 K).
2.8. ADME Prediction
SWISSADME online (http://www.swissadme.ch) [53] and ProTox-3.0 online software (https://tox.charite.de/protox3/index.php?site) [54] prediction tools were used to evaluate the physicochemical properties, lipophilicity, water solubility, pharmacokinetics, drug-likeness and toxicity prediction, violations of Veber’s rule [55], violations of Lipinski’s rule of five [56] only one violation is accepted in case of variables [57].
3. RESULTS AND DISCUSSION
3.1. Flax Microgreens and Growth Conditions
The flaxseed germination time was about 8 days under controlled conditions. Figure 2 presents well-grown flax microgreens in a highly controlled environment called “Hi-Tech Polyhouse” at LPU. Following their growth, extraction of phytochemicals from flax microgreens using methanol showed a good yield (33%).
3.2. Qualitative Screening Tests of the Methanolic Extract of Flax Microgreens
The qualitative analysis of the methanolic extract of flax microgreens showed the presence of numerous phytochemical constituents such as alkaloids, saponins, flavonoids, steroid, cardiac glycoside, coumarins, phenolic compounds, and chalcones whereas Tannins, Terpenoids, Emodins were absent as shown in Table S1 (see supplementary material), which agrees to the finding of Monica & Joseph [58] and Hanaa et al. [59] of about 80%. This can be attributed due to the high content of phytochemicals present microgreens.
3.3. GC-MS Profiling of Methanolic Extract of Flax Microgreens
A total of 58 chromatogram peaks were identified from methanolic extract of flax microgreens which corresponding to the bioactive compounds and were recognized by relating their retention time, peak area (%), peak height (%), and mass spectral fragmentation patterns to that of the known compounds described by the NIST library. According to research conducted by Farag et al. [60], 28 phytochemicals were identified from flaxseed whereas the current study revealed that 58 different phytocompounds were identified from flax microgreens, this clearly indicated that flax microgreens are rich in phytocompounds. The total ion chromatogram is presented in Figure 3, Table S2 (see supplementary material) presents the phytocompounds along with their corresponding molecular formula, retention time, MW, and peak area (%).
All phytocompounds were docked against the target proteins and their binding affinities were recorded (See supplementary material). Among those, two lignans, i.e., CID11002708 and CID290541 exhibited the highest binding affinity and convenient molecular interaction across the target receptors. Figure 4 presents the ion chromatogram of the two phytochemicals, CID11002708 and CID290541, which exhibited the highest binding affinities to PCa target proteins.
 | Figure 2. Well grown flax microgreens under controlled conditions in Hi-Tech polyhouse facility. [Click here to view] |
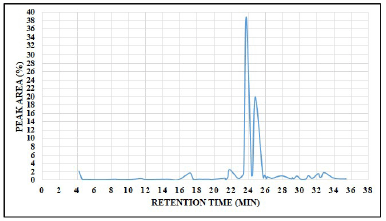 | Figure 3. GC-MS chromatogram of methanolic extract of flax microgreens (Linum usitatissimum L.). [Click here to view] |
 | Figure 4. (a) (3R,4R)-3,4-bis(1,3-benzodioxol-5-ylmethyl)oxolan-2-one and (b) 5-[4-(1,3-benzodioxol-5-yl)-2,3-dimethylbutyl]-1,3-benzodioxole. [Click here to view] |
 | Figure 5. Three-dimensional structure of drug targets, docking procedures, and ADME properties. [Click here to view] |
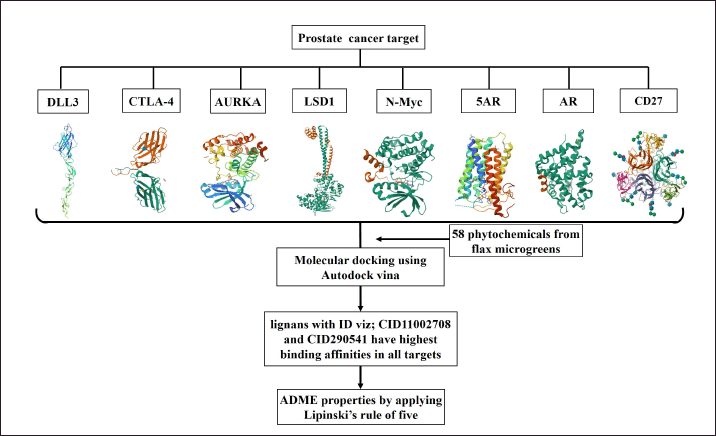
3.4. Prostate Cancer Target Proteins
It may be possible to stop or slow the spread of PCa to other areas of the body by targeting a particular protein that is frequently overexpressed in the disease. The important PCa target proteins include Aurora A kinase (AURKA) [61], DLL3 [62], N-myc proto-oncogene protein (N-Myc) [63], Cytotoxic T-lymphocyte antigen 4 (CTLA-4) [64], 5α-Reductase (5AR) [65–68], Androgen receptor (AR) [69], Lysine-specific histone demethylase 1A (LSD1) [70], and CD27 [71]. The structure of all the target proteins except DLL3 was available and downloaded from PDB (http://www.rscb.org/pdb). Therefore, the modeling studies, sequence analysis, selection, and validation of DLL3 were done using different bioinformatic tools. The three-dimensional structure of drug targets, docking procedures, and ADME properties are depicted in Figure 5.
3.5. Protein Modeling Studies and Sequence Analysis of DLL3
The structure of DLL3 is not available on PDB database. Therefore, The DLL3 sequence of humans was retrieved in FASTA format from the protein sequence and functional information database (UniprotKB) with sequence identity (ID: Q9NYJ7). The ExPASy ProtParam server was used to compute the physicochemical properties of the query sequence and tabulated in Table 1. The result shows isoelectric point (pI), AI, Instability Index (II) and GRAVY values of query protein DLL3 were 7.590, 66.540, 53.730, and −0.1870, respectively, which corroborate with the findings of Joshi et al. [72]. The isoelectric point (pI) shows the acidity or basicity nature of the protein. With a wider temperature range, the query protein’s greater AI demonstrates its stability. The hydrophilic nature of the protein is indicated by the negative GRAVY values for protein.
The InterPro and CDD of the NCBI were used to confirm the protein domains and functional sites after they were identified using the Prosite database. Tables 2 and 3 present the domain, profile, and patterns found for DLL3’s characteristic functionalities. Six conserved EGF-like domains were found by both Prosite, InterPro, and CDD, which is in line with the DLL3’s function and the body of existing literatures. Table 3 illustrates the amino acid sequences for each of the six EGF 3 domains, along with their respective disulfide bond positions. Figure 6 presents the sequence logo which helps in visualizing the cysteine residue position for disulfide bond in the EGF domain.
 | Table 1. The physicochemical parameters of DLL3. [Click here to view] |
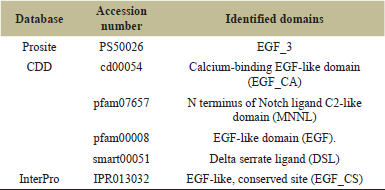 | Table 2. Identified domains from the sequence. [Click here to view] |
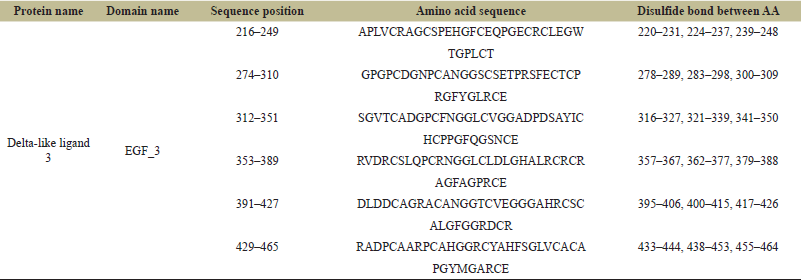 | Table 3. Prosite result shows the structure, sequence position and disulfide bond between amino acids in domains. [Click here to view] |
 | Figure 6. Sequence Logo for DLL-3 domain. [Click here to view] |
 | Figure 7. Transmembrane helix prediction of DLL-3 using DeepTMHMM (1.0.24). [Click here to view] |
PSSpred and SOPMA online tools were used to study and analyze the structural features of the DLL3 as shown in Table S3 and Figure S1 (see supplementary material). The result of SOPMA is comparable with that of PSSpred prediction. The secondary structure of DLL3 shows the extended strands, beta turns, alpha helix, and domination of random coils. The PredictProtein server shows a very high percentage in turns and coils, whereas in strands and helix was significantly lower. According to the region’s solvent accessibility, 45.31%, 41.10%, and 13.59% of DLL3’s regions are buried, exposed to solvent, and intermediate regions, respectively. The result from DeepTMHMM 1.0.24 revealed that the predicted amino acids in signal, extracellular, transmembrane, and cytoplasmic regions were 1–26, 27–491, 492–513, and 514–618, respectively, (Figure 7 and Table 4) which agrees with the findings of Joshi et al. [72] of about 70%. This can be attributed due to DeepTMHMM 1.0.24 server updation.
3.6. Homology Modeling and Validation
Figure 8 presents the accurate prediction of the C2, DSL, and EGF domains generated from the SWISS-MODEL server. Newly modeled protein subjected to additional analysis viz; Ramachandran plot analysis, distance plot standard deviation, RMSD values, secondary structure, cytoplasmic and transmembrane helix prediction. Figure 8 presents the sequence alignment between Human DLL3 and Delta-like protein 1(DLL1). The template used for modeling of DLL3 protein was the crystal structure of Delta-like ligand (DLL1) with PDB ID (4XBM). The reliability and quality of the generated model were determined. Figure 9 presents the homology-modeled structure of Human DLL3. The quality of the built protein was further evaluated using PROVE and PROCHECK server. Protein residues are categorized using the Ramachandran plot according to their areas in the quadra plot and the φ and ψ angles of the protein backbone. Glycine is represented by triangles in the quadra plot, while other amino acids are represented by squares. The yellow and red areas represent the allowed and most allowed regions, respectively. The Ramachandran plot of both (query and template sequences) of protein which has been generated using SWISS MODEL is shown in Figure 10. The residues in black color showed the most favored and allowed regions, whereas the red color indicates either the residues are in disallowed or allowed regions. Table 5 shows that 0.6% of amino acid residues were in the disallowed region. Figure S2 (see supplementary material) presents ERRAT plot in chains A and B of modeled DLL3 protein. The ERRAT plot also shows some of the regions with high error, these results suggest the need for model refinement. After completion of three iterations of loop refinement, there is no residue in the ERRAT plot displays a high error and ~99% residues are present in generously allowed regions in the plot. The atomic calculation in the form of z-score from PROVE server indicates the quality of the modeled protein structure (Table 6).
 | Table 4. Prediction of the DLL3 domain locations using DeepTMHMM 1.0.24. [Click here to view] |
3.7. Molecular Docking Analysis
Eight target proteins were downloaded from the Protein Databank and using grid box analysis, the binding pocket was manually predicted [46]. The phytocompounds in .sdf format were obtained from the PubChem database and converted into .pdbqt format [44]. The PyRx tool, a virtual screening tool that uses both Vina and Autodock, was used for molecular docking [43]. The binding affinities of 58 flax microgreen phytocompounds against PCa target proteins are shown in Table S4 (see supplemental material), from which the best two lignans (CID290541 and CID11002708) were chosen for further study. Table 7 presents the docking result of two lignans against eight PCa target proteins. The binding affinities ranged from −5.3 to −10.6 Kcal/mol across the docking result. The results indicate that CID11002708 shows the best binding affinity to several target proteins compared to CID290541. CID11002708 exhibited strong binding affinity (−6.60, −7.90, −9.50, −10.20, and −8.50 kcal/mol) toward CTLA-4, AURKA, 5AR, AR, and CD27, respectively, making it a more useful candidate for multi-target therapy. Compared to CID11002708, CID29054 was less effective generally across the different protein targets, although having a slightly higher binding affinity (−5.80 and −10.60 kcal/mol) toward DLL3 and LSD1. The docking results were also compared to some of the FDA-approved drugs for the PCa such as Mitoxantrone chloride (CID51082), Flutamide (CID3397), and Darolutamide (CID67171867) [73]. The binding affinity of FDA-approved drugs were presented in Table 8. As compared to standard drugs, the CID11002708 and CID290548 showed binding affinities closer to that of FDA-approved drugs and can be consider as novel inhibitors for the treatment of PCa. According to the findings of Joshi et al. [72], the binding energy of DLL3 and reference inhibitor Nicotine is −5.9 kcal/mol, this aligned with current findings of the studied phytochemicals (CID11002708 and CID290541) have binding affinities of −5.3 and −5.8 kcal/mol against DLL3, respectively. CID11002708 demonstrated a binding affinity of −7.9 kcal/mol against AR, which is comparable to or better than known AR inhibitors such as enzalutamide [74]. Similarly, CID11002708’s binding affinity of −10.2 kcal/mol for 5AR is higher than that of finasteride’s binding affinity of −9.8 kcal/mol [75], and it could serve as a novel candidate in this category of PCa inhibitors.
 | Figure 8. Aligment between human DLL3 and DLL1. [Click here to view] |
 | Figure 9. Homology modeled structure of human DLL3. [Click here to view] |
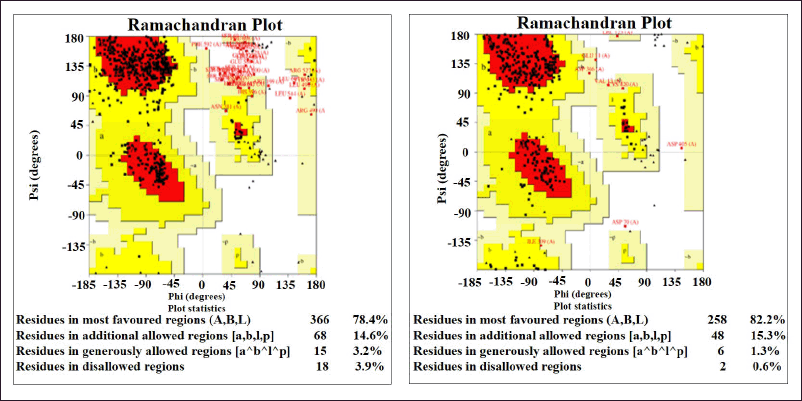 | Figure 10. Ramachandran map of Q9NYJ7 (Query sequence) and template sequence model. [Click here to view] |
 | Table 5. PROCHECK tool generates a Ramachandran plot for the final DLL3 models. [Click here to view] |
3.8. Inhibition Constant (Ki) Prediction
To understand the anti-cancer activity of the presently studied compounds, the Inhibition constant (Ki) value predictions were done. The inhibition constant (Ki) value is a useful parameter to quantitatively measure the concentration of compound required to produce half maximum inhibition to a given biological process and is universally used to symbolize the inhibitory effect of compounds [76]. Table 9 shows the predicted Inhibition constant (Ki) value for the studied compounds (CID11002708 and CID29054) were in a range of 0.0175–129.750 micro molar. The CID11002708 has shown the best possible inhibitory potential with 0.0175 micro molar, whereas CID29054 has shown the least predicted Inhibition constant (Ki) value across the protein targets. In comparison to control FDA drugs, the CID11002708 and CID29054 show the predicted Inhibition constant (Ki) value closer to that of control FDA drugs.
 | Table 6. PROVE analysis for the DLL3 model. [Click here to view] |
 | Table 7. Molecular docking result of flax microgreens lignans (CID11002708 and CID290541) against prostate cancer target proteins. [Click here to view] |
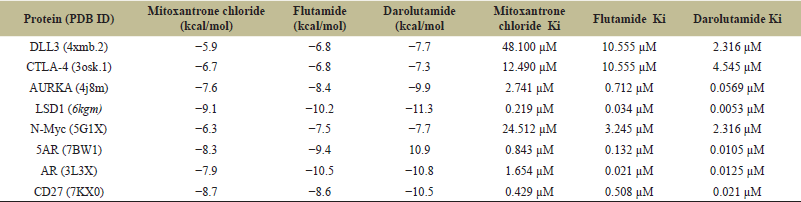 | Table 8. Binding affinities and inhibition constant (Ki) values of some of the FDA approved drugs across eight target proteins. [Click here to view] |
 | Table 9. Physicochemical properties, lipophilicity and drug-likeness of CID290541 and CID11002708. [Click here to view] |
 | Figure 11. 3D Docking pose interactions of DLL3 protein with (a) 290541 (b) 11002708. [Click here to view] |
 | Figure 12. 3D Docking pose interactions of CTLA-4 protein with (a) 290541 (b) 11002708. [Click here to view] |
 | Figure 13. 3D Docking pose interactions of AURKA protein with (a) 290541 (b) 11002708. [Click here to view] |
3.9. Protein–Ligand Interactions of CID11002708 and CID290541 Against PCa Target Proteins
Understanding the mechanism of action through protein-ligand interactions is highly important. These interactions involve different forces which include; electrostatic, hydrogen bonding, and hydrophobic interactions. The binding affinity alone cannot give positive results for protein-ligand binding strength and stability [77]. However, considering the amino acid residues involved in the protein-ligand interaction will support the docking result, this can also increase the docking result’s credibility. The results show that the amino acid residues favorably interact with CID11002708 and CID290541 compounds at the target proteins’ active sites. Hydrogen bonds and hydrophobic interactions show that these ligands are positively interact with the binding site of the enzyme, this could possibly lead to enzyme inhibition which is necessary in drug design by targeting the specific receptor (Table 7). The protein-ligand interaction stabilized the ligands to perfectly fit into the binding pocket of the target proteins. 2D representation is illustrated in Figure S3−S10 (see supplementary material) (LIGPLOT+ software version 2.2) and 3D representation is illustrated in Figures 11−18 (PLIP online server) [78].
 | Figure 14. 3D Docking pose interactions of LSD1 `protein with (a) 290541 (b) 11002708. [Click here to view] |
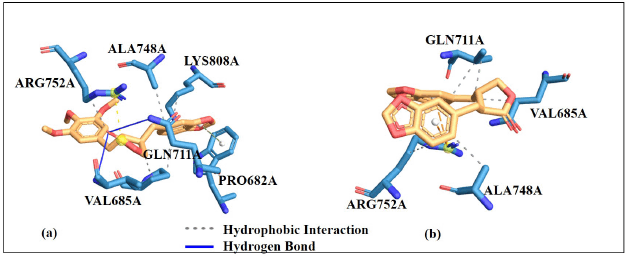 | Figure 15. 3D Docking pose interactions of androgen receptor protein with (a) 290541 (b) 11002708. [Click here to view] |
 | Figure 16. 3D Docking pose interactions of N-Myc protein with (a) 290541 (b) 11002708. [Click here to view] |
 | Figure 17. 3D Docking pose interactions of 5AR protein with (a) 290541 (b) 11002708. [Click here to view] |
 | Figure 18. 3D Docking pose interactions of CD27 protein with (a) 290541 (b) 11002708. [Click here to view] |
 | Table 10. Toxicity prediction of CID290541 and CID11002708 using ProTox-3.0 online tool. [Click here to view] |
3.10. ADME/T Properties Prediction
ProTox-3.0 (https://tox.charite.de/protox3/index.php?site) and SWISSADME (http://www.swissadme.ch/) online tools were used to get the ADMET (Absorption, Distribution, Metabolism, Excretion, and Toxicity) properties. Acute oral toxicity, blood-brain barrier (BBB), carcinogenicity, nutritional toxicity, hepatotoxicity, nephrotoxicity, neurotoxicity, cytochrome P450 inhibitors isoforms (CYP inhibitors), hepatotoxicity, human ether-a-go-go-related gene inhibition (hERG), human intestinal absorption, human oral bioavailability, and P-glycoprotein inhibitor are among the ADMET parameters assessed for the compounds (Tables 9 and 10). Table 9 presents the results that show that the compounds have good BBB permeation, high gastrointestinal absorption, and good oral bioavailability in humans. Lipinski’s rule of five and Veber’s filter were used to study the bioavailability of CID290541 and CID11002708. According to Lipinski’s rule of five, compounds with an octanol/water partition coefficient (LogPo/w) of less than five, an MW of less than 500, less than ten H-bond acceptors, and less than five H-bond donors were predicted to exhibit favorable bioavailability [56]. Additional parameters were expanded by the Veber rule to include a topological polar surface area with values of 79.89–109.35 (preferably TPSA ≤140 Å2) and rotatable bonds (preferably n-ROTB < 10) [55]. The Egan rule considered good bioavailability for compounds with (TPSA ≤132 Å2 and -1 < LogP < 6) [79]. Both the studied compounds obeyed Lipinski’s rule of five as well as Veber’s filter and exhibited favorable bioavailability.
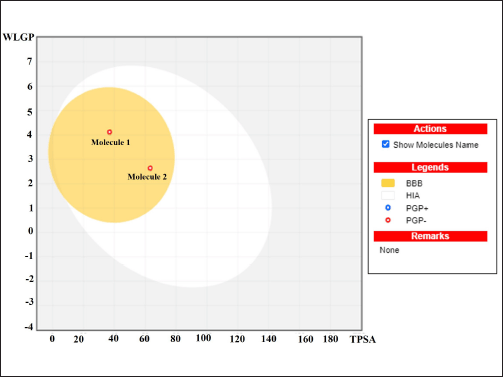 | Figure 19. The Boiled-egg’s yolk depicted molecule 1 (290541) and molecule 2 (11002708). [Click here to view] |
According to the toxicity prediction study (Table 10), the compounds CID290541 and CID11002708, which are classified as Classes IV and V, respectively, show no acute oral toxicity. This indicates that the compounds are not digested in the gastrointestinal tract before reaching the intended target and have reduced oral toxicity [80]. Furthermore, the compounds are non-hepatotoxic, non-nutritional toxic, non-nephrotoxic, non-neurotoxic, and non-inhibitor of hERG. In terms of predicting the efflux by P-glycoprotein from the cell, the compounds are non-inhibitors and non-substrate of permeability glycoprotein. If a drug is an inhibitor of P-glycoprotein, it will inhibit the efflux process from the cell and increase bioavailability; if it is not an inhibitor of P-glycoprotein, it will efflux from the cell by P-glycoprotein, limit bioavailability by pumping back into the lumen and possibly encourage the drug’s excretion into the urine and bile [81]. In the case of metabolism, the compounds are inhibitors to most CYP450 isoforms with the exception of CYP2E1. A non-inhibitor of cytochrome P450 means that the molecule will not hinder the biotransformation of the compound (drug) metabolized by cytochrome P4 [82].
The result from Figure 19 presents a boiled egg’s yolk graph of compounds, both molecule 1 (CID290541) and molecule 2 (CID11002708) are in the yellow section which clearly indicated that they can passively be absorbed and penetrate the BBB.
4. CONCLUSION
In conclusion, this study revealed that flax microgreens are a rich source of essential fatty acids and lignans. The findings of this study suggest that lignans from flax microgreens, particularly CID11002708 and CID290541, could serve as promising candidates for PCa therapy. These compounds exhibited significant binding affinities and favorable interactions with cancer-related proteins, supporting their potential use in drug development. The toxicity prediction study demonstrated that the compounds have low toxicity and specific metabolic characteristics. However, further in vitro and in vivo studies are necessary to validate these docking results and fully understand the therapeutic potential and safety of these compounds.
5. AUTHOR CONTRIBUTION
All authors contributed to the study conception and design. ML contributed to the formal analysis of the experiment, methodology, visualization, and writing-original draft. AKV and GR extensively edited the manuscript. AKV helped in computational research work. NRS participated in a critical review. GR helped supervise the project and gave final approval for the version to be published. All authors have read and agreed to the published version of the manuscript.
6. ACKNOWLEDGMENT
The authors extend their appreciation to the Deanship of School of Bioengineering and Biosciences, Lovely Professional University, Phagwara, Punjab, India.
7. CONFLICTS OF INTEREST
The authors declare no conflict of interest.
8. AUTHORS’ DECLARATION
The authors hereby declare that this article is original and that any liability for claims relating to the content of this article will be borne by them.
9. ETHICAL APPROVALS
This study does not involve experiments on animals or human subjects.
10. DATA AVAILABILITY
All the data is available with the authors and shall be provided upon request.
11. PUBLISHER’S NOTE
All claims expressed in this article are solely those of the authors and do not necessarily represent those of the publisher, the editors and the reviewers. This journal remains neutral with regard to jurisdictional claims in published institutional affiliation.
12. USE OF ARTIFICIAL INTELLIGENCE (AI)-ASSISTED TECHNOLOGY
The authors declares that they have not used artificial intelligence (AI)-tools for writing and editing of the manuscript, and no images were manipulated using AI.
REFERENCES
1. Mazurakova A, Samec M, Koklesova L, Biringer K, Kudela E, Al-Ishaq RK, et al. Anti-prostate cancer protection and therapy in the framework of predictive, preventive and personalised medicine—comprehensive effects of phytochemicals in primary, secondary and tertiary care. EPMA J 2022 Sep;13(3):461–86.
2. Packer JR, Maitland NJ. The molecular and cellular origin of human prostate cancer. Biochimica et Biophysica Acta (BBA)-Mol Cell Res 2016 Jun 1;1863(6):1238–60.
3. Nnadi CO, Okorie NH, Nwodo NJ. Evaluation of In Vitro antiprotozoal and cytotoxic activities of selected medicinal plants used in Nigerian folk medicine. TJNPR 2021 Apr 1;5(4):609–12.
4. Matowa PR, Gundidza M, Gwanzura L, Nhachi CF. A survey of ethnomedicinal plants used to treat cancer by traditional medicine practitioners in Zimbabwe. BMC Complementary Med Therap 2020 Dec;20:1–3.
5. Stavropoulos P, Mavroeidis A, Papadopoulos G, Roussis I, Bilalis D, Kakabouki I. On the path towards a “greener” EU: a mini review on flax (Linum usitatissimum L.) as a case study. Plants 2023 Mar 1;12(5):1102.
6. Morris DH. Flax: A health and nutrition primer. 4th edition, Flax Council of Canada, Saskatoon, Canada, 2007. Available via www.flaxcouncil.ca
7. Adolphe JL, Whiting SJ, Juurlink BH, Thorpe LU, Alcorn J. Health effects with consumption of the flax lignan secoisolariciresinol diglucoside. Br J Nutr 2010 Apr;103(7):929–38.
8. Pan A, Sun J, Chen Y, Ye X, Li H, Yu Z, et al. Effects of a flaxseed-derived lignan supplement in type 2 diabetic patients: a randomized, double-blind, cross-over trial. PLoS One 2007 Nov 7;2(11):e1148.
9. Chiang YM, Liu HK, Lo JM, Chien SC, Chan YF, Lee TH, et al. Cytotoxic constituents of the leaves of Calocedrus formosana. J Chinese Chem Soc 2003 Feb;50(1):161–6.
10. Sliwoski G, Kothiwale S, Meiler J, Lowe EW. Computational methods in drug discovery. Pharmacol Re 2014 Jan 1;66(1):334–95.
11. Demark-Wahnefried W, Polascik TJ, George SL, Switzer BR, Madden JF, Ruffin IV MT, et al. Flaxseed supplementation (not dietary fat restriction) reduces prostate cancer proliferation rates in men presurgery. Cancer Epidemiol Biomarkers Prev 2008 Dec 1;17(12):3577–87.
12. Johnsson P. Bioactive phytochemicals in flaxseed. Acta Uni Agric Suec 2009.
13. Saggar JK, Chen J, Corey P, Thompson LU. Dietary flaxseed lignan or oil combined with tamoxifen treatment affects MCF-7 tumor growth through estrogen receptor-and growth factor-signaling pathways. Mol Nutr Food Res 2010 Mar;54(3):415–25.
14. Viveky N, Thorpe L, Alcorn J, Hadjistavropoulos T, Whiting S. Safety evaluation of flaxseed lignan supplementation in older adults residing in long-term care homes. Nurs Home Res 2015;1:84–8.
15. Lopez J, Tait SW. Mitochondrial apoptosis: killing cancer using the enemy within. Br J Cancer 2015 Mar;112(6):957–62.
16. Zaman S, Wang R, Gandhi V. Targeting the apoptosis pathway in hematologic malignancies. Leuk Lymphoma. 2014 Sep 1;55(9):1980–92.
17. Hassan M, Watari H, AbuAlmaaty A, Ohba Y, Sakuragi N. Apoptosis and molecular targeting therapy in cancer. BioMed Res Int 2014 Oct;2014:150845.
18. Jang WY, Kim MY, Cho JY. Antioxidant, anti-inflammatory, anti-menopausal, and anti-cancer effects of lignans and their metabolites. Int J Mol Sci 2022 Dec 7;23(24):15482.
19. Otto T, Sicinski P. Cell cycle proteins as promising targets in cancer therapy. Nat Rev Cancer. 2017 Feb;17(2):93–115.
20. Diaz-Moralli S, Tarrado-Castellarnau M, Miranda A, Cascante M. Targeting cell cycle regulation in cancer therapy. Pharmacol Therap 2013 May 1;138(2):255–71.
21. Vargas-Madriz ÁF, Kuri-García A, Vargas-Madriz H, Chávez-Servín JL, Ferriz-Martínez RA, Hernández-Sandoval LG, et al. Phenolic profile and antioxidant capacity of Pithecellobium dulce (Roxb) Benth: a review. J Food Sci Technol 2020 Dec;57:4316–36.
22. Velavan S. Phytochemical techniques-a review. World J Sci Res 2015;1(2):80–91.
23. Rajasree RS, Ittiyavirah SP, Naseef PP, Kuruniyan MS, Anisree GS, Elayadeth-Meethal M. An evaluation of the antioxidant activity of a methanolic extract of Cucumis melo L. fruit (F1 hybrid). Separations 2021 Aug 18;8(8):123.
24. NIST Chemistry Web Book. In: Mallard WG, Linstrom PJ (Editors). NIST Standard Reference Database. National Institute of Standards and Technology, Gaithersburg, MD, 2008. Available via https://webbook.nist.gov.
25. UniProt Consortium. UniProt: a worldwide hub of protein knowledge. Nucleic Acids Res 2019 Jan 8;47(D1):D506–15.
26. Guruprasad K, Reddy BB, Pandit MW. Correlation between stability of a protein and its dipeptide composition: a novel approach for predicting in vivo stability of a protein from its primary sequence. Protein Eng Des Sel 1990 Dec 1;4(2):155–61.
27. Kyte J. A simple method for displaying the hydropathic character of a protein. J Mol Biol 1993;268:10558–63.
28. Gill SC, Von Hippel PH. Calculation of protein extinction coefficients from amino acid sequence data. Anal Biochem 1989 Nov 1;182(2):319–26.
29. Ikai A. Thermostability and aliphatic index of globular proteins. J Biochem 1980 Oct 1;88(6):1895–8.
30. Gasteiger E, Hoogland C, Gattiker A, Duvaud SE, Wilkins MR, Appel RD, et al. Protein identification and analysis tools on the ExPASy server. Humana Press, New York, NY, 2005.
31. Walker JM, editor. The proteomics protocols handbook. Humana Press, New York, NY, pp 571–607, 2005.
32. Sigrist CJ, De Castro E, Cerutti L, Cuche BA, Hulo N, Bridge A, et al. New and continuing developments at PROSITE. Nucleic Acids Res 2012 Nov 17;41(D1):D344–7.
33. Lu S, Wang J, Chitsaz F, Derbyshire MK, Geer RC, Gonzales NR, et al. CDD/SPARCLE: the conserved domain database in 2020. Nucleic Acids Res 2020 Jan 8;48(D1):D265–8.
34. Combet C, Blanchet C, Geourjon C, Deleage G. NPS@: network protein sequence analysis. Trends Biochem Sci 2000 Mar 1;25(3):147–50.
35. Yan R, Xu D, Yang J, Walker S, Zhang Y. A comparative assessment and analysis of 20 representative sequence alignment methods for protein structure prediction. Sci Rep 2013 Sep 10;3(1):2619.
36. Krogh A, Larsson B, Von Heijne G, Sonnhammer EL. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J Mol Biol 2001 Jan 19;305(3):567–80.
37. Yachdav G, Kloppmann E, Kajan L, Hecht M, Goldberg T, Hamp T, et al. PredictProtein—an open resource for online prediction of protein structural and functional features. Nucleic Acids Res 2014 Jul 1;42(W1):W337–43.
38. Waterhouse A, Bertoni M, Bienert S, Studer G, Tauriello G, Gumienny R, et al. SWISS-MODEL: homology modeling of protein structures and complexes. Nucleic Acids Res 2018 Jul 2;46(W1):W296–303.
39. Ramachandran GN, Ramakrishnan C, Sasisekharan V. Stereochemistry of polypeptide chain configurations. J Mol Biol 1963 Jul;7:95–9.
40. Baseer AQ, Mushfiq S, Monib AW, Hassand MH, Niazi P. Computational structural analysis and homology modelling of beta-xylanase from Bifidobacterium pullorum: a comprehensive in-silico investigation. J Res Appl Sci Biotechnol 2023 Dec 16;2(6):49–57.
41. Colovos C, Yeates TO. Verification of protein structures: patterns of nonbonded atomic interactions. Protein Sci 1993 Sep;2(9):1511–9.
42. Lüthy R, Bowie JU, Eisenberg D. Assessment of protein models with three-dimensional profiles. Nature 1992 Mar 5;356(6364):83–5.
43. Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, et al. UCSF Chimera—a visualization system for exploratory research and analysis. J Comput Chem 2004 Oct;25(13):1605–12.
44. Verma AK, Gulati P, Lakshmi GB, Solanki PR, Kumar A. Interaction studies of Gut metabolite; trimethylene amine oxide with bovine serum albumin through spectroscopic, DFT and molecular docking approach. bioRxiv. 2023 Apr 6:2023–04.
45. Dallakyan S, Olson AJ. Small-molecule library screening by docking with PyRx. Chem Biol Methods Protoc 2015;1263:243–50.
46. Lawal M, Verma AK, Umar IA, Gadanya AM, Umar B, Yahaya N, et al. Analysis of new potent anti-diabetic molecules from phytochemicals of Pistia strateotes with Sglt1 and G6pc proteins of Homo Sapiens for treatment of diabetes mellitus. An in silico approach. Silico Approach IOSR JPBS 2020;15:59–73.
47. Gulati P, Kumar Verma A, Kumar A, Solanki P. Para-Cresyl sulfate and BSA conjugation for developing aptasensor: spectroscopic methods and molecular simulation. ECS J Solid State Sci Technol 2023 Jul 12;12(7):073004.
48. Verma AK, Sharma S, Jayaraj A, Deep S. In silico study of interaction of (ZnO) 12 nanocluster to glucose oxidase-FAD in absence and presence of glucose. J Biomol Struct Dyn 2023 Dec 29;41(24):15234–42.
49. Spackman PR, Turner MJ, McKinnon JJ, Wolff SK, Grimwood DJ, Jayatilaka D, et al. CrystalExplorer: a program for Hirshfeld surface analysis, visualization and quantitative analysis of molecular crystals. J Appl Crystallogr 2021 Jun 1;54(3):1006–11.
50. Kumar S, Mohan A, Sharma NR, Kumar A, Girdhar M, Malik T, et al. Computational frontiers in aptamer-based nanomedicine for precision therapeutics: a comprehensive review. ACS Omega 2024 Jun 10;9(25):26838–62.
51. Verma AK, Mishra A, Dhiman TK, Sardar M, Solanki PR. Experimental and in silico interaction studies of alpha amylase-silver nanoparticle: a nano-bio-conjugate. bioRxiv 2022 Jun 13:2022–06.
52. Ortiz CL, Completo GC, Nacario RC, Nellas RB. Potential inhibitors of galactofuranosyltransferase 2 (GlfT2): molecular docking, 3D-QSAR, and in silico ADMETox studies. Sci Rep 2019 Nov 19;9(1):17096.
53. Daina A, Michielin O, Zoete V. SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci Rep 2017 Mar 3;7(1):42717.
54. Banerjee P, Kemmler E, Dunkel M, Preissner R. ProTox 3.0: a webserver for the prediction of toxicity of chemicals. Nucleic Acids Res 2024;52:W513–20.
55. Veber DF, Johnson SR, Cheng HY, Smith BR, Ward KW, Kopple KD. Molecular properties that influence the oral bioavailability of drug candidates. J Med Chem 2002 Jun 6;45(12):2615–23.
56. Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev 2012 Dec 1;64:4–17.
57. Kaur B, Rolta R, Salaria D, Kumar B, Fadare OA, da Costa RA, et al. An in-silico investigation to explore anti-cancer potential of Foeniculum vulgare Mill. Phytoconstituents for the management of human breast cancer. Molecules 2022 Jun 24;27(13):4077.
58. Monica SJ, Joseph M. Phytochemical screening of flaxseed (Linum usitatissimum L.). Int J Sci Res 2016 Mar;5(3):218–20.
59. Hanaa MH, Ismail HA, Mahmoud ME, Ibrahim HM. Antioxidant activity and phytochemical analysis of flaxseeds (Linum usitatisimum L.). Minia J Agric Res Dev 2017 Nov;37(1):129–40.
60. Farag SM, Essa EE, Alharbi SA, Alfarraj S, El-Hassan GA. Agro-waste derived compounds (flax and black seed peels): toxicological effect against the West Nile virus vector, Culex pipiens L. with special reference to GC–MS analysis. Saudi J Biol Sci 2021 Sep 1;28(9):5261–7.
61. Otto T, Horn S, Brockmann M, Eilers U, Schüttrumpf L, Popov N, et al. Stabilization of N-Myc is a critical function of Aurora A in human neuroblastoma. Cancer Cell 2009 Jan 6;15(1):67–78.
62. Rudin CM, Pietanza MC, Bauer TM, Ready N, Morgensztern D, Glisson BS, et al. Rovalpituzumab tesirine, a DLL3-targeted antibody-drug conjugate, in recurrent small-cell lung cancer: a first-in-human, first-in-class, open-label, phase 1 study. Lancet Oncol 2017 Jan 1;18(1):42–51.
63. Gustafson WC, Meyerowitz JG, Nekritz EA, Chen J, Benes C, Charron E, et al. Drugging MYCN through an allosteric transition in Aurora kinase A. Cancer Cell 2014 Sep 8;26(3):414–27.
64. Yu C, Sonnen AF, George R, Dessailly BH, Stagg LJ, Evans EJ, et al. Rigid-body ligand recognition drives cytotoxic T-lymphocyte antigen 4 (CTLA-4) receptor triggering. J Biol Chem 2011 Feb 25;286(8):6685–96.
65. Robitaille J, Langlois VS. Consequences of steroid-5α-reductase deficiency and inhibition in vertebrates. Gen Comp Endocrinol 2020 May 1;290:113400.
66. Marks LS. 5α-reductase: history and clinical importance. Rev Urol 2004;6(Suppl 9):S11.
67. Schmidt LJ, Tindall DJ. Steroid 5 α-reductase inhibitors targeting BPH and prostate cancer. The J Steroid Biochem Mol Biol 2011 May 1;125(1-2):32–8.
68. Aggarwal S, Thareja S, Verma A, Bhardwaj TR, Kumar M. An overview on 5α-reductase inhibitors. Steroids 2010 Feb 1;75(2):109–53.
69. Liss MA, Thompson IM. Prostate cancer prevention with 5-alpha reductase inhibitors: concepts and controversies. Curr Opin Urol 2018 Jan 1;28(1):42–5.
70. Niwa H, Sato S, Handa N, Sengoku T, Umehara T, Yokoyama S. Development and structural evaluation of N-alkylated Trans-2-phenylcyclopropylamine-based LSD1 inhibitors. Chem Med Chem 2020 May 6;15(9):787–93.
71. Liu W, Maben Z, Wang C, Lindquist KC, Li M, Rayannavar V, et al. Structural delineation and phase-dependent activation of the costimulatory CD27: CD70 complex. J Biol Chem 2021 Oct 1;297(4):101102.
72. Joshi BP, Bhandare VV, Patel P, Sharma A, Patel R, Krishnamurthy R. Molecular modeling studies and identification of novel phytochemical inhibitor of DLL3. J Biomol Struct Dyn 2023 May 3;41(7):3089–109.
73. https://www.cancer.gov/about-cancer/treatment/drugs/prostate
74. Ito Y, Sadar MD. Enzalutamide and blocking androgen receptor in advanced prostate cancer: lessons learnt from the history of drug development of antiandrogens. Res Rep Urol 2018 Feb 16:23–32.
75. Saah SA, Sakyi PO, Adu-Poku D, Boadi NO, Djan G, Amponsah D, et al. Docking and molecular dynamics identify leads against 5 alpha reductase 2 for benign prostate hyperplasia treatment. J Chem 2023;2023(1):8880213.
76. Rao CM, Yejella RP, Rehman RS, Basha SH. Molecular docking based screening of novel designed chalcone series of compounds for their anti-cancer activity targeting EGFR kinase domain. Bioinformation 2015;11(7):322.
77. Cheng J, Hao Y, Shi Q, Hou G, Wang Y, Wang Y, et al. Discovery of novel chinese medicine compounds targeting 3CL protease by virtual screening and molecular dynamics simulation. Molecules 2023 Jan 17;28(3):937.
78. Laskowski RA, Swindells MB. LigPlot+: multiple ligand–protein interaction diagrams for drug discovery. J Chem Inf Model 2011;51(10):2778–86.
79. Egan WJ, Merz KM, Baldwin JJ. Prediction of drug absorption using multivariate statistics. J Med Chem 2000 Oct 19;43(21):3867–77.
80. Srivastava V, Yadav A, Sarkar P. Molecular docking and ADMET study of bioactive compounds of Glycyrrhiza glabra against main protease of SARS-CoV2. Mat Today Proc 2022 Jan 1;49:2999–3007.
81. Finch A, Pillans P. P-glycoprotein and its role in drug-drug interactions. Aust Prescr 2014 Aug 4;37(4):137–9.
82. Cheng F, Li W, Zhou Y, Shen J, Wu Z, Liu G, et al. admetSAR: a comprehensive source and free tool for assessment of chemical ADMET properties. J Chem Inf Model 2012 Nov 26;52(11):3099–105.