
1. INTRODUCTION
The structures of protein complexes are basics for understanding the sub-atomic systems of protein–protein interaction, analyzing genetic disorders, and controlling binding affinity. The difficulties with respect to time and cost for X ray-crystallography and nuclear magnetic resonance methodologies create a gap between the experimentally determined complex structures and sequences accessible for protein buildings [1]. The inadequacy of complex structures can be mitigated by molecular docking, which gives a quick and proficient alternative in the field of bioinformatics.
Molecular Docking is a tool to predict the energetically favored conversion of one molecule to the next molecule when bound to form a relatively stable complex with the overall least energy. Docking expects to accomplish an optimized conformation and relative direction between protein–ligand. It helps us to see how small ligands bind to different proteins, for example, transport protein, signal receptors, catalysts, viral proteins, and so forth so as to suppress or actuate their function. For molecular docking, it is basic to recognize the ligands that can bind protein at an explicit dynamic site.
Docking is utilized in in-silico screening of huge databases of compounds for hit ID and assessing the impacts of chemical alterations during lead optimization [2,3]. Hit identification is the significant objective of high throughput screening (HTS) that intends to recognize dynamic compounds (hits) by screening enormous quantities of various chemical compounds against chosen targets [4]. For most of the disease-related proteins, experimental structures are not accessible thus; ligand docking can be performed on hypothetical models [5]. The inaccuracies in a homology modeling can be overcome by utilizing computational docking techniques which decide the gross basic highlights of a complex, but find it exceedingly difficult to effectively anticipate high-goals structures of such protein–protein complex, indicating the need to form new docking calculations which include the essential degrees of freedom to make up for the errors.
Thus, the present study intends to portray the leading edge of protein–ligand docking and its application looking for appropriate antisickling agents from Carica papaya.
2. DOCKING PROCEDURE
Whole docking procedure can be described in following simple steps:
2.1. Search for Protein and Ligand Structure
The search for convenient target protein and ligands is essential for performing docking. So, one has to search whether the target protein is deposited in the Protein Data Bank (PDB) database (http://pdb.org) or Swiss UniProt knowledge base (http://expasy.org/sprot). If the target protein is not present in the database but similar sequences are there, then homology modeling can be done by using the Swiss model repository (http://swissmodel.expasy.org/repository/), modeller tools––I-TASSER, etc. Next, the ligand can be found from the PubChem database (http://pubchem.org) or Zinc (http://blaster.docking.org/zinc/).
2.2. Search for Protein–Ligand Binding Site
The search for potential protein–ligand binding site can be achieved by using two approaches, i.e., (a) first, identify the protein–ligand binding site and then dock the ligand (b) dock ligand directly onto the complete receptor structure; blind docking [6].
2.3. Development of Docking Program
Molecular docking has turned into an essential part in many drug discovery programs, particularly for virtual screening of phytochemicals as potential drug molecules. The first docking software was developed in the mid-1980s by Irwin Kuntz, University of California and attempts are still in the process to improve the docking calculations. Ongoing advancements in docking tools predict the capacity of an enzyme by distinguishing its natural substrates [7]. Table 1 gives the characteristic features of some important docking programs.
 | Table 1: Key attributes of some chosen protein–ligand docking programs. [Click here to view] |
The successful predictions of protein complexes were done by identifying the protein of interest belonging to a specific super family so that the search for potential substrates and types of reactions are restricted to a specific area. However, various methodologies were utilized to precisely rank the docked molecules by using different programs such as:
- DOCK 3.5.x––This program used the idea that enzymes catalyze reactions by restricting the transition state superior to the substrate, docking transition state-like molecules ought to give a superior signal than docking substrates by using the knowledge of amidohydrolase super family performed hydrolysis reactions. The protein was kept rigid [8].
- Glide––The program distinguishes the enzymes having a place with a specific subgroup of enolase super family permitted tapering the arrangement of potential substrates, and precision in the positioning was improved by refining and rescoring the docked complex with an increasingly confounded material science-based scoring capacity and permitting receptor side chains to move [9].
2.4. Mechanism of Docking
2.4.1. Search algorithms
The principle target of the search algorithm is to locate every single imaginable direction and conformation of the protein combined with the ligand. It is classified into two types-
- Systematic methods or Direct methods––There are three subtypes of systematic methods as follows:
- Conformational search––Here, the structural parameter of ligand, i.e., torsional (dihedral) translational and rotational degrees of freedom are altered gradually [10].
- Fragmentation––Multiple fragments are either dock and tried to interface them with bonds, or anchor fragments is dock first and manufacture outward in ventures from that bound position. Tools––Flex XTM, LUDI, DOCK, etc.
- Database search––Various sensible conformations of each and every small molecule that is already stored in the database can be generated by this method and then dock them as firm bodies. Tools––FLOG
- Stochastic methods or Random methods––There are three subtypes of stochastic methods.
- Monte Carlo (MC)––In this method, ligands are haphazardly set in the receptor binding site, it is then scored, and the new configuration is created by haphazard changes. Tools––ICM, MCDOCK, etc.
- Genetic algorithm (GA)––It begins with a populace of postures, where descriptors of configuration and position in respect to the receptor are the “gene” and the score is the “fitness”; perform transformations, hybrids, etc., of the fittest to make the next generation and rehash to concurrence [11]. Tools––AutoDock, GOLD, etc.
- Tabu search (TS)––It works by striking restrictions that prevent the previously exposed territories of the ligands conformational space from being checked again and, therefore, support the study of a novel configuration. Tools––PRO_LEADS, Molegro Virtual Docker (MVD)TM.
2.4.2. Scoring functions
Scoring functions helps in the evaluation of which ligand configuration and rotation is most favorable with respect to the receptor (protein) and by using virtual screening ranked them (ligand) according to their binding affinity [12]. The scoring function is categorized into four major groups:
- Force field-based––It measures the binding affinity by adding the contribution of bond-like angle bonding, and torsional deviation and non-bonded interaction like van der Waal forces, hydrogen holding, or Columbic electrostatics in an ace capacity [13]. Tools—GoldScore, AutoDock, DOCK, etc.
- Empirical function––It depends on multiple linear relapse investigation of a preparation set of complex structures by utilizing protein–ligand complex with known binding affinities, containing functional groups and sort of interaction. For example, N-O hydrogen bond, O-O hydrogen bond, salt scaffold, aromatic ring stacking, etc. [14]. Tools—LUDI score, ChemScore, AutoDock scoring, etc.
- Knowledge-based––It gives separation ward pair potential to particles, functional groups by statistical analyzing of a set of complex structures [15]. Tools––PMF, DrugScore, etc.
- Consensus––It basically consolidates the scores or rankings got from various scoring capacities in different ways.
2.5. Software’s for Docking
There are various softwares used for docking and some of the important ones are mentioned as follows:
2.5.1. Dock (http://dock.compbio.ucsf.edu/.)
The Dock is a structure-based design developed in the 1980s by Irwin Kuntz and co-worker, University of California. Firstly Dock version 1 (Dock 1) calculated the quality of ligand and receptor complex on steric overlap [16]. Later on, Dock 2 provides more sophisticated control on the sampling algorithm, which provides better timing and accuracy [17].
Dock 3 includes an extremely thorough and careful parameter for an input of ligand and receptors. The first virtual screening of enzyme thymidylate synthase was performed by using Dock 3 [18]. Dock 4 was developed with an improved graph matching algorithm for ligand orientation [19]. Dock 6 (version 6.0–6.8) is being used for performing a docking with improved sampling, scoring, optimization, and numerous bug fixes [20]. Latest version Dock 6.9 provides new ligand searching method DOCK_DN, i.e., De Novo design using program based assembly [21].
2.5.2. Autodock (http://autodock.scripps.edu)
AutoDock is a broadly utilized non-business docking program. It was the first docking programming that ties ligand with full compliance adaptability. The Autodock was created by Prof. Arthur J. Olson at the Department of molecular biology of The Scripps Research Institute in La Jolla (CA, USA). The software comprises two consecutively connected programs, i.e., AutoDock and AutoGrid [22].
AutoGrid is utilized to compute the non-covalent vitality of interaction between the firm piece of the receptor and a probe atom that is available in the ligands that will be docked for the receptor. The principle job of AutoDock is to control the docking procedure of the chosen ligand through the cross section volume.
There are several versions of AutoDock like AutoDockFR that re-enacts fractional receptor adaptability by permitting countless unequivocally indicated receptor side-chains to investigate their conformational space while scanning for vivaciously positive restricting postures for a given ligand. Researcher made higher docking progress rates by utilizing receptor adaptability in the binding site of receptor adaptations that are tentatively decided without the ligand present, for example, Apo conformations [23].
AutoDock Vina can achieve two orders of magnitude speedup in comparison to AutoDock 4. With the help of AutoDock Vina, accuracy level increases twice as compared to previously published software and naturally ascertains the framework maps and bunches the outcomes in a manner straightforward to the client [24].
2.5.3. Argus lab 4.0.1 (http://www.arguslab.com)
Argus lab is a molecular modeling software developed by Mark Thomson, Department of Energy at Pacific Northwest National Laboratory, USA. Argus lab is based on algorithms for modeling solvent effects by combining quantum mechanics with classical mechanics. This program can be used for drug designing, generating graphics, and molecular modeling.
2.5.4. Genetic optimization for ligand docking (GOLDTM) (http://www.ccdc.cam.ac.uk/products/life_sciences/gold)
GOLD's principle qualities are that: (i) spine and side chain adaptability can be incorporated into the computations, (ii) the program utilizes client characterized scoring capacities and can adjust likewise, (iii) the vitality capacities depend on both conformational and non-reinforced contact data. (iv) an assortment of imperative alternatives can be characterized for the docking, (v) crystallographic water particles in the ligand binding site can be well-out during the docking, (vi) it can handles metal atoms consequently on the off chance that they are set up effectively in the protein information record, and (vii) virtual screening high throughput screening results can be broke down and post-prepared effectively with the sidekick programs SILVERTM or GoldMineTM.
Latest version of GOLD Suite 5.2 is a combination of three components, i.e., Gold 5.2 to carry out protein–ligand docking, Hermes 1.6 for comprehensive protein visualisation, and Gold Mine 1.5 for analysis of docking grades.
2.5.5. MVD (Molecule & allegro virtual docker)
MVDTM (MolDock) is developed by Molegro Aps [25]. MVD's special qualities are: (i) it automatically allocates charges, bond orders, hybridization, and add hydrogen to the given structures, (ii) it naturally foresees potential binding destinations in the receptor, (iii) it manages receptor side chain adaptability by considering incited fit interaction, (iv) it docks in predetermined vitality lattices (which accelerates the figuring’s), (v) it manages client characterized imperatives during docking, (vi) it can profit by the utilization of layouts (for example, pharmacophores) during docking, and (vii) it can convey the computations on various PCs.
2.6. Applications of Molecular Docking
- Modeling the structure of a protein–ligand complex is significant for understanding the coupling interaction between a potential restorative compound (the ligand) and its remedial objective (the protein).
- The motion space of the protein–ligand complex can be explored by using computer-aided docking so as to compute a vigorously steady configuration that models the structure of the complex [26].
- The investigation of the movement space is finished by an inspecting calculation and the steadiness of confirmation of the complex is assessed by utilizing a scoring or vitality work that measures the binding affinity of the complex.
- Drug configuration dependent on the peptides or peptidomimetics is quickly picking up footing in the pharmaceutical business. These compounds are getting to be prominent as a result of their low lethality and high explicitness. Enthusiasm for these compounds has likewise expanded with the advancement of refined assembling methods. The quantity of peptides approved by the United States Food and Drug Administration is expanding at a yearly pace of 8% and it is anticipated that the market for peptide-based medications will be tremendous.
- In expansion to pharmacodynamics information, e.g., strength, proclivity, viability, selectivity, pharmacokinetic properties, ADMET: absorption, distribution, metabolism, excretion, and toxicity have likewise been concentrated through the use of these techniques.
- Covalent medications have exhibited to be lucky options in a few remedial zones, for example, malignant growth, diabetes, and contagious, cardiovascular, gastrointestinal and neurologic ailments. Recent studies suggest that around 33% of enzyme modulators present in the market are covalent inhibitors. Covalent ligands irreversibly inactivate their target due to which recuperation of the biological functions requires the re-synthesis of the target molecule [27].
3. MOLECULAR DOCKING AS A TOOL FOR IDENTIFYING POTENTIAL DRUGS TO PREVENT SICKLE CELL ANEMIA
Sickle cell anemia is an autosomal recessive hereditary disease caused due to alteration in hemoglobin gene due to which the red blood cells (RBCs) become sickle/bow-shaped rather than round-shaped (Fig. 1) [28]. Sickle cell disease is known to affect the population living mostly in Africa, India, the Mediterranean, and Middle Eastern [29,30]. Blood of sickle cell patients contains a surprisingly enormous number of juvenile cells and many long, lean, bow-shaped erythrocytes that look like the sharp edge of a sickle. At the point when hemoglobin from sickle cells (Hbs) is deoxygenated, it brings about polymerization and misshapes the RBCs into a sickle shape [31].
 | Figure 1: Difference between normal and sickle red blood cells. [Click here to view] |
Nucleotide sequencing of β-globin mRNA from the sickle β-globin gene uncovered that the typical codon GAG at position β6 has been replaced by GUG [32]. This change decides the addition of valine at this position rather than glutamic acid as in the case of normal HbA [33]. The sickness and characteristic appear in individuals of African lineage, Mediterranean nations, India, and the Middle-East however once in a while in individuals of European and white lineage also. People suffer from many health problems such as anemia, bacterial infection, brain damage, thrombosis, liver damage, strokes, etc. [34].
3.1. Antisickling Property in Plants
Various plant species show antioxidant treatment vital for the counteractive action of thick cell arrangement and free radicals preventing oxidative cell damage leading to prolongation of red blood cell life [35]. Table 2 enlists some of the important plants used in cure and prevention of sickle cell disease.
3.2 Carica papaya Linn.
Carica papaya is a long herbaceous plant, with plentiful smooth latex coming to 16–20 ft. in stature, the stem up to 30 cm thick, evergreen, erect, a perennial plant in habitat (Fig. 2). Carica papaya Linn. belongs to family Caricaceae which is known for its medicinal property like antimicrobial, antibacterial, antiulcer, anticancer, antioxidant, and antifungal. Papaya is being used since the past time for treating jaundice and sickle cell disease by the tribes of Nigeria. Many scientists reported that ripe fruit and leaf of C. papaya are used for treating sickle cell in humans [42–44].
3.3. Sickle Cell Target Protein (2HBS Protein)
A computational approach for drug discovery is done by the protein–ligand docking, in which an active site of a protein is taken as the target and inhibited by a ligand that may be a synthetic or a natural compound as a drug. There are several well-known target sites for drug-resistant treatment/control. Here, the target protein is employed for the evaluation and effectiveness of plant chemical compound obtained from C. papaya.
 | Figure 2: Carica papaya plant. [Click here to view] |
.png) | Table 2: Phytocompound used in managing sickle cell disease and its modes of action [36]. [Click here to view] |
4. METHOD FOR DOCKING OF SICKLE CELL PROTEIN WITH AN ANTISICKLING AGENT
The discussed antisickling compounds and target protein were taken for docking study; these were downloaded from PubChem and PDB. Downloaded 3D protein structure and compound's relevant information are listed in Table 3. Furthermore, conversion of the (.sdf) file to the (.pdb) file of phyto-compounds was performed in online SMILE translator and the removal of the water molecule and other ligands from target protein was done by Discovery Studio Visualizer 16.1.0 before docking attempts. Argus lab 4.0 was used for docking attempts, and PyMol v1.6 and Discovery Studio Visualizer 16.1.0 software were used for their interaction study.
 | Table 3: Name of the phytochemicals with their 3D structure and docking scores against sickle cell protein (2HBS). [Click here to view] |
4.1. Target Protein Selection for Docking Attempts
The sickle cell protein, 2 Deoxyhemoglobin S (2HBS) (Fig. 3) was downloaded from Protein Data Bank and phytochemicals were taken from the PubChem database. PyMOL and Discovery studio software is used for analysis and interaction study.
4.2. Phytocompound Selection for Docking Attempts
Carica species contains various secondary metabolites including cyanogenic glycosides, amines and alkaloids, gums, fatty acids, terpenes (counting basic oils, phytosterol, triterpene genins, diterpenes, and saponins), nonprotein amino acids, hydrolyzable tannins, consolidated tannins, and flavonoids. The plant is more extravagant in glycine, carpaine, anthraquinone, phenylalanine, tryptophan, and tyrosine. The plant chemical compounds like N-methyl aspartic acid, oleic acid, Hexasiloxane dodecamethyl-, 2-Methoxy-4-vinylphenol, and Cyclohexyl isothiocyanate, D-Glucitol, Hexaacetate, Phenylalanin amide, and Papaverinol, etc., are found in leaf and unripe fruits. Papaya latex contains four cysteine compounds like papain, chymopapain, glycyl endopeptidase, and caracain. Reports are there on the impact of amino acids on gelatine kinetics and solubility of sickle cells [45]. Aromatic amino acids like tyrosine, tryptophan, and phenylalanine were significantly more energetic as anti-sickling [46]. The dense cell formation due to polymerization of the sickle cell can be inhibited by using nutritional antioxidants in combination with Vit. C and E [47]. Many workers have also reported beta carotene, Vit. C, gamma terpinene, citric acid, methionine, lycopene, tartaric acid, alanine, and sucrose have antisickling properties [48].
4.3. Docking with 2HBS
For this study, Argus lab 4.0 was used to dock ligand (phytochemicals from C. papaya) into the active site of 2HBS protein. Argus lab has been reported as an effective software having the ability to predict bound configuration and binding energies of the ligand with macromolecular targets rapidly and precisely. Polar hydrogen atoms were added to the ligands and its non-polar hydrogen particles were combined. All the adaptable bonds were set to be adaptable. Protein–ligand docking was finished utilizing the GA strategy. The grid box with a dimension of 20 × 20 × 25 and 0.400 Å frameworks separating was to cover the protein binding site and suit ligands to move unreservedly. Subsequent to docking searches were finished, the best configuration was looked for the most populated bunch with the minimum binding vitality. The connection of docked protein–ligand complex configuration, including hydrogen bond and different interactions were broke down utilizing Discovery Studio Visualizer 16.1.0.
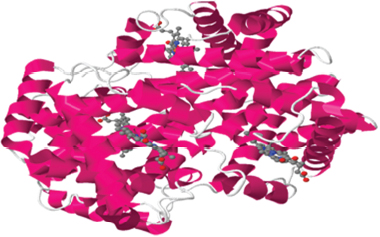 | Figure 3: Sickle cell protein 2HBS (PDB no.-10.2210/pdb2hbs/pdb). [Click here to view] |
4.4 Steps Involved in Docking of 2HBS
4.4.1. Downloading of protein structures and ligands
- The sickle cell protein (2HBS) was downloaded from protein data bank using protein id and opened in the Argus Lab program. The molecule Tree view tool of 2HBS (positioned on the left part of the monitor) was expanded, plus the residue/misc folder was opened up. The selection of various ligands was taken from previously reviewed articles and then downloaded from Pubchem sites in .sdf format. Ligands were converted to .PDB format using an online smile translator.
4.4.2. Creation of ligand and binding site groups
- Sickle cell protein inhibitor was selected from “1440 LK2” in the Tree View, which appeared yellow.
- The Edit/Hide Unselected menu choice was chosen to conceal all molecules that are not chosen. The only atoms appearing on the screen ought to be the sickle cell protein inhibitor.
- Center the sickle cell protein inhibitor in the window by choosing the View/Centre Molecule in the Window menu on the toolbar.
- While the inhibitor is chosen, hydrogen atoms were included by squeezing the H key on the toolbar.
- Right side click on the “1440 LK2” in the Tree View and the “Make a Ligand Group from this Residue” choice was chosen. Argus lab will build a cluster underneath the Groups file with a similar name “1 LK2” that is of sort Ligand.
- Left side clicks on either “1440 LK2” residue in the Residues/Misc organizer. This activity will again choose the atoms of the Ligand on the monitor.
- The selected residue was copied and pasted. In the Residues/Misc organizer in the Tree View tool, a recently featured residue with a name “2480 LK2” will be observed.
- Ligand cluster of this new residue was made in a similar way as done beforehand. Right-tapped on “2480 LK2” and “Make a Ligand Group from this Residue” was selected.
- The binding site for the ligand cluster by right clicking on the ligand cluster in the Groups folder and select the “Make a Binding Site Group for this Group” menu choice. This will create a Binding Site that comprises all residues that have at least one atom within 3.5Å from any atom in the ligand cluster.
4.4.3. Docking the ligand into the definite binding site
- Dock Setting dialog box was brought up by choosing the Calculation/Dock a Ligand menu choice on the toolbar.
- The ligand to dock in the “Ligand” drop-box was chosen. Make a point to choose the cluster named “ligand” group.
- The “Calculate Size” key was clicked and a docking box custom made to the binding site was made which appeared on the monitor.
- Made sure that “GA Dock” is the chosen docking the Calculation type is “Dock” and the Ligand is Flexible.
- Docking calculation was started by clicking on “Start” button.
- Various docking scores were shown in the screen but the best score was taken into consideration.
5. DOCKING RESULT
The results were based on free energy binding and the lowest binding energy. The phytocompounds showing the most negative binding energies were considered to have the strongest binding affinity toward sickle cell protein (2HBS). Twenty-four phytocompounds from C. papaya (Table 3) are docked against sickle cell protein, i.e., 2HBS from which three compounds showed the best results. These compounds were Xanthoangelol D; N-[(4R)-4-(3-fluorophenyl)-6-oxo-4,5-dihydro-H-pyrimidin-2-yl]-3-methoxy benzamide, and Carpaine showing minimum energy values in protein–ligand interaction against sickle cell protein (2HBS). The docking of Xanthoangelol D formed a stable complex by the formation of hydrogen bond with ALA1126, GLU1004, ALA1125, PRO1122, PRO893, HIS856, LYS896, ALA217, HIS218, SER905, ASP220, LEU219, LEU904, LEU890, and PHE889 with a binding free energy of −10.5994 kcal/mol, N-[(4R)-4-(3-fluorophenyl)-6-oxo-4,5-dihydro-H-pyrimidin-2-yl]-3-methoxybenzamide formed hydrogen bond with GLY224, PHE226, THR228, THR225, PRO1122, ALA1007, VAL1003, GLU1004, THR1001, ASP214, LYS896, HIS856, LEU890, VAL1123, ALA1126, and HIS218 with an energy of −9.33596 kcal/mol, whereas that of Carpaine to 2HBS showed THR225, ASP214, ALA1126, THR1001, PRO1122, ASP220, ASN221, HIS906, LYS896, HIS856, DEF1152, and ALA217 establishing a hydrogen bond with Carpaine with a binding energy of −9.23601 kcal/mol (Fig. 4). Several workers have used docking methods in search of phytocompounds for possible treatment of diseases or degradation of environmental pollutants. Kurjogi et al. [49] used the docking method in search of possible natural drug for Staphylococcus aureus enterotoxins (SEs) like SEA and SEB and based on the minimum binding energy concluded that 28-Norolean-12-en-3-one, a compound present in flowering plants of Lardizabalaceae family acts as a good inhibitor for SEA, while Betulin, a natural triterpene isolated from bark of birch trees will act as good inhibitor for SEB-like enterotoxins. Satapute et al. [50] docked triazole fungicide propiconazole against the superoxide dismutase (SOD) and catalase enzyme of plasmid cured Pseudomonas aeruginosa and found that propiconazole binds more efficiently with catalase as compared to SOD and can degrade propiconazole.
 | Figure 4: Interactions of the ligand (a) Xanthoangelol D, (b) N-[(4R)-4-(3-fluorophenyl)-6-oxo-4,5-dihydro-H-pyrimidin-2-yl]-3-methoxybenzamide, and (c) Carpaine with binding site residues of sickle cell protein (PDB ID:2HBS). [Click here to view] |
6. Conclusions
Docking studies were carried out in order to find the inhibitory activity of the compounds found in leaf extract and ripe fruit of C. papaya. Among the 24, three compounds showed the best docking result based on the binding energy. Docking studies and binding free energy calculations of these 24 compounds revealed that Xanthoangelol D has minimum interaction energy (−10.5994 kcal/mol), followed by N-[(4R)-4-(3-fluorophenyl)-6-oxo-4,5-dihydro-H-pyrimidin-2-yl]-3-methoxybenzamide (−9.33596kcal/mol), and Carpaine (−9.23601kcal/mol). Hence, the given phytocompounds with minimum binding energy can be further used in a wet lab for the production of drugs to cure or inhibit the action of sickle cell protein.
The current review has highlighted the principle and methods by which molecular docking has been applied in the identification of novel phytocompounds yet challenges still remain, particularly with exactness and scoring capacity governed by quantum mechanics. Most docking program effectively predicts the binding methods of small particles ligands with receptor binding destinations. The present algorithm does not measure the complete energy related with intermolecular interactions with adequate precision. However, in the present panorama of medication revelation, where high steady loss rates are a noteworthy concern, properly structured virtual screening procedures are efficient, financially savvy, and beneficial options. With the help of molecular docking, we can identify promising phytochemicals that can be used as future medicine for curing diseases.
From the above the experiment, it is concluded that Sickle cell anemia has been a major health concern worldwide and has proven to claim the lives of many people yearly [51]. Till now, no cure has been successfully developed for this genetic disease. The current study was focused on an assessment of the inhibitory activity of leaf extract and ripe fruit compounds of C. papaya leaves against sickle cell protein. Among the identified 24 compounds, three compounds [Xanthoangelol D,N-[(4R)-4-(3-fluorophenyl)-6-oxo-4, 5-dihydro-H-pyrimidin-2-yl]-3-methoxy benzamide, and Carpaine] showed better binding affinity toward the sickle cell protein (2HBS). Different modes of interaction such as hydrogen bonding and other hydrophobic interactions were observed between the ligands and the 2HBS protein of sickle cell anemia. The information acquired through this study on the binding mode of phytocompounds from C. papaya and 2HBS protein will highly facilitate the synthesis and testing of these compounds as drugs for sickle cell disease. The study suggested that compounds from the C. papaya will be potent drug candidates against sickle cell anemia.
ACKNOWLEDGMENT
The authors DRD, DK, and BPD are grateful to the DBT-BIF (Govt. of India) for funding and the authorities of the Department of Biosciences and Biotechnology, Fakir Mohan University, Balasore for providing laboratory facilities and encouragement. DK acknowledges UGC DS Kothari post doctoral scheme for his fellowship.
AUTHOR’S CONTRIBUTION
DK and BPD developed the concept. DRD, DK, and PK wrote the manuscript.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
REFERENCES
1. Gohlke H, Klebe G. Approaches to the description and prediction of the binding affinity of small-molecule ligands to macromolecular receptors. Angew Chem Int Ed 2002;41(15):2644–76. CrossRef
2. Kitchen DB, Decornez H, Furr JR, Bajorath J. Docking and scoring in virtual screening for drug discovery: methods and applications. Nat Rev Drug Discov 2004;3(11):935. CrossRef
3. Warren GL, Andrews CW, Capelli AM, Clarke B, LaLonde J, Lambert MH, et al. A critical assessment of docking programs and scoring functions. J Med Chem 2006;49(20):5912–31. CrossRef
4. Shun TY, Lazo JS, Sharlow ER, Johnston PA. Identifying actives from HTS data sets: practical approaches for the selection of an appropriate HTS data-processing method and quality control review. J Biomol Screen 2011;16(1):1–4. CrossRef
5. Bordogna A, Pandini A, Bonati L. Predicting the accuracy of protein-ligand docking on homology models. J Comput Chem 2011;32(1):81–98. CrossRef
6. Pujadas G, Vaque M, Ardevol A, Blade C, Salvado MJ, Blay M, et al. Protein-ligand docking: a review of recent advances and future perspectives. Curr Pharm Anal 2008;4(1):1–9. CrossRef
7. Kuntz ID, Blaney JM, Oatley SJ, Langridge R, Ferrin TE. A geometric approach to macromolecule-ligand interactions. J Mol Biol 1982;161(2):269–88. CrossRef
8. Hermann JC, Marti-Arbona R, Fedorov AA, Fedorov E, Almo SC, Shoichet BK, et al. Structure-based activity prediction for an enzyme of unknown function. Nature 2007;448(7155):775. CrossRef
9. Kalyanaraman C, Imker HJ, Fedorov AA, Fedorov EV, Glasner ME, Babbitt PC, et al. Discovery of a dipeptide epimerase enzymatic function guided by homology modeling and virtual screening. Structure 2008;16(11):1668–77. CrossRef
10. Sousa SF, Fernandes PA, Ramos MJ. Protein–ligand docking: current status and future challenges. Proteins Struct Funct Bioinformatics 2006;65(1):15–26. CrossRef
11. Jones G, Willett P, Glen RC, Leach AR, Taylor R. Development and validation of a genetic algorithm for flexible docking. J Mol Biol 1997;267(3):727–48. CrossRef
12. Huang SY, Grinter SZ, Zou X. Scoring functions and their evaluation methods for protein–ligand docking: recent advances and future directions. Phys Chem Chem Phys 2010;12(40):12899–908. CrossRef
13. Englebienne P, Moitessier N. Docking ligands into flexible and solvated macromolecules. 5. Force-field-based prediction of binding affinities of ligands to proteins. J Chem Inf Model 2009;49(11):2564–71. CrossRef
14. Murray CW, Auton TR, Eldridge MD. Empirical scoring functions. II. The testing of an empirical scoring function for the prediction of ligand-receptor binding affinities and the use of Bayesian regression to improve the quality of the model. J Comput Aided Mol Des 1998;12(5):503–19. CrossRef
15. Huang SY, Zou X. An iterative knowledge-based scoring function to predict protein–ligand interactions: I. Derivation of interaction potentials. J Comput Chem 2006;27(15):1866–75. CrossRef
16. DesJarlais RL, Sheridan RP, Seibel GL, Dixon JS, Kuntz ID, Venkataraghavan R. Using shape complementarity as an initial screen in designing ligands for a receptor binding site of known three-dimensional structure. J Med Chem 1988;31(4):722–9. CrossRef
17. Shoichet BK, Kuntz ID. Protein docking and complementarity. J Mol Biol 1991;221(1):327–46. CrossRef
18. Goodford PJ. A computational procedure for determining energetically favorable binding sites on biologically important macromolecules. J Med Chem 1985;28(7):849–57. CrossRef
19. Ewing TJ, Makino S, Skillman AG, Kuntz ID. DOCK 4.0: search strategies for automated molecular docking of flexible molecule databases. J Comput Aided Mol Des 2001;15(5):411–28. CrossRef
20. Wang J, Wolf RM, Caldwell JW, Kollman PA, Case DA. Development and testing of a general amber force field. J Comput Chem 2004;25(9):1157–74. CrossRef
21. Allen WJ, Fochtman BC, Balius TE, Rizzo RC. Customizable de novo design strategies for DOCK: application to HIVgp41 and other therapeutic targets. J Comput Chem 2017;38(30):2641–63. CrossRef
22. Morris GM, Goodsell DS, Huey R, Olson AJ. Distributed automated docking of flexible ligands to proteins: parallel applications of AutoDock 2.4. J Comput Aided Mol Des 1996;10(4):293–304. CrossRef
23. Ravindranath PA, Forli S, Goodsell DS, Olson AJ, Sanner MF. AutoDockFR: advances in protein-ligand docking with explicitly specified binding site flexibility. PLoS Comput Biol 2015;11(12):1004586. CrossRef
24. Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem 2010;31(2):455–61. CrossRef
25. Thomsen R, Christensen MH. MolDock: a new technique for high-accuracy molecular docking. J Med Chem 2006;49(11):3315–21. CrossRef
26. Dhanik A, McMurray JS, Kavraki LE. DINC: a new AutoDock-based protocol for docking large ligands. BMC Struct Biol 2013;13(1):S11. CrossRef
27. Ferreira L, dos Santos R, Oliva G, Andricopulo A. Molecular docking and structure-based drug design strategies. Molecules 2015;20(7):13384–421. CrossRef
28. Bunn HF. Pathogenesis and treatment of sickle cell disease. N Engl J Med 1997;337(11):762–9. CrossRef
29. Bownas J. Genetic profile: sickle cell anemia. National Institutes of Health Publication, Maryland, pp 96–4057, 2000.
30. Buchanan GR, DeBaun MR, Quinn CT, Steinberg MH. Sickle cell disease. ASH Educ Program Book 2004;2004(1):35–47. CrossRef
31. Pauling L, Itano HA, Singer SJ, Wells IC. Sickle cell anemia, a molecular disease. Science 1949;110(2865):543–8. CrossRef
32. Marotta CA, Wilson JT, Forget BG, Weissman SM. Human beta-globin messenger RNA. III. Nucleotide sequences derived from complementary DNA. J Biol Chem 1977;252(14):5040–53.
33. Perutz MF, Lehmann H. Molecular pathology of human haemoglobin. Nature 1968;219(5157):902. CrossRef
34. Enang U. Current concepts in the management of sickle cell disorder (a practical guide). Kraft Books Ltd., Ibadan, Nigeria, pp 41–45, 1992.
35. Tatum VL, Chow CK. Antioxidant status and susceptibility of sickle erythrocytes to oxidative and osmotic stress. Free Radic Res 1996;25(2):133–9. CrossRef
36. Ameh SJ, Tarfa FD, Ebeshi BU. Traditional herbal management of sickle cell anemia: lessons from Nigeria. Anemia 2012; 2012:1–9. CrossRef
37. Thomas KD, Ajani B. Antisickling agent in an extract of unripe pawpaw fruit (Carica papaya). Trans R Soc Trop Med Hyg 1987;81(3):510–1. CrossRef
38. Mpiana PT, Mudogo V, Kabangu YF, Tshibangu DS, Ngbolua KN, Atibu EK, et al. Antisickling activity and thermostability of anthocyanins extract from a congolese plant, Hymenocardia acida Tul. (Hymenocardiaceae). Int J Pharmacol 2009;5(1):65–70. CrossRef
39. Ekeke GI, Shode FO. Phenylalanine is the predominant antisickling agent in Cajanus cajan seed extract. Planta Med 1990;56(01):41–3. CrossRef
40. Fall AB, Vanhaelen-Fastré R, Vanhaelen M, Lo I, Toppet M, Ferster A, et al. In vitro antisickling activity of a rearranged limonoid isolated from Khaya senegalensis. Planta Med 1999;65(03):209–12. CrossRef
41. Ameh SJ, Obodozie OO, Inyang US, Abubakar MS, Garba M. Climbing black pepper (Piper guineense) seeds as an antisickling remedy. InNuts and seeds in health and disease prevention. Academic Press, Cambridge, MA, pp 333–43, 2011. CrossRef
42. Oduola T, Adeniyi FA, Ogunyemi EO, Bello IS, Idowu TO. Antisickling agent in an extract of unripe pawpaw (Carica papaya): is it real?. Afr J Biotechnol 2006;5(20):1947–9.
43. Mojisola OC, Adebolu EA, Alani DM. Antisickling properties of Carica papaya Linn. J Nat Prod 2008;1:56–8.
44. Imaga NO, Gbenle GO, Okochi VI, Akanbi SO, Edeoghon SO, Oigbochie V, et al. Antisickling property of Carica papaya leaf extract. Afr J Biochem Res. 2009;3(4):102–6.
45. Noguchi CT, Schechter AN. Effects of amino acids on gelation kinetics and solubility of sickle hemoglobin. Biochem Biophys Res Commun 1977;74(2):637–42. CrossRef
46. Iqbal T, Kazi GH. Amino acid composition of protein hydrolysates of some lentils of common use. J Chem Soc Pak 1980;2:61–6.
47. Ohnishi ST, Ohnishi T. In vitro effects of aged garlic extract and other nutritional supplements on sickle erythrocytes. J Nutr2001;131(3):1085S–92S. CrossRef
48. Duke JA. Database of phytochemical constituents of GRAS herbs and other economic plants. CRC Press, Washington DC, 1992.
49. Kurjogi M, Satapute P, Jogaiah S, Abdelrahman M, Daddam J, Ramu V, et al. Computational modeling of the staphylococcal enterotoxins and their interaction with natural antitoxin compounds. Int J Mol Sci 2018;19(1):133–45. CrossRef
50. Satapute P, Sanakal RD, Mulla SI, Kaliwal B. Molecular interaction of the triazole fungicide propiconazole with homology modelled superoxide dismutase and catalase. Environ Sustainabil 2019;1–11. CrossRef
51. Dash BP, Archana Y, Satapathy N, Naik SK. Search for antisickling agents from plants. Pharmacogn Rev 2013;7(13):53–60. CrossRef