1. Wettstein DV, Mikhaylenko G, Froseth JA, Kannangara CG. Improved barley broiler feed with transgenic malt containing heat-stable (1,3-1,4)-b-glucanase. Proc Natl Acad Sci USA 2000;97:13512-7. [CrossRef]
2. Lamp AE, Evans AM, Moritz JS. The effects of pelleting and glucanase supplementation in hulled barley based diets on feed manufacture, broiler performance, and digesta viscosity. J Appl Poult Res 2015;24:295-303. [CrossRef]
3. Bull AT, Chesters CG. The biochemistry of laminarin and the nature of laminarinase. Adv Enzymol 1963;28:325. [CrossRef]
4. Mann JW, Jeffries TW, Macmillan JD. Production and ecological significance of yeast cell wall degrading enzymes from Oerskovia. Appl Environ Microbiol 1978;36:594. [CrossRef]
5. Blackman LM, Cullerne DP, Hardham AR. Bioinformatic characterisation of genes encoding cell wall degrading enzymes in the Phytophthora parasitica genome. BMC Genomics 2014;15:785. [CrossRef]
6. Cantarel BL, Coutinho PM, Rancurel C, Bernard T, Lombard V, Henrissat B. The carbohydrate-active enzymes database (CAZy): An expert resource for glycogenomics. Nucleic Acids Res 2009;37:233-8. [CrossRef]
7. Bielecki S, Galas E. Microbial beta-glucanases different from cellulases. Crit Rev Biotechnol 1991;10:275-304. [CrossRef]
8. Bayat A. Science, medicine, and the future: Bioinformatics. BMJ 2002;324:1018-22. [CrossRef]
9. McGregor N, Morar M, Fenger TH, Stogios P, Lenfant N, Yin V, et al. Structure-function analysis of a mixed-linkage β-glucanase/xyloglucanase from the Key ruminal Bacteroidetes Prevotella bryantii B(1)4. J Biol Chem 2016;291:1175-97. [CrossRef]
10. Lim J, Lee C, Dhakshnamoorthy V, Park JS, Hong S. Molecular characterization of Streptomyces coelicolor A(3) SCO6548 as a cellulose 1,4-b-cellobiosidase. FEMS Microbiol Lett 2016;363:245. [CrossRef]
11. Lanka S, Talluri VR, Ganesh J, Latha NL. Homology modeling, molecular dynamic simulations and docking studies of a new cold active extracellular lipase, EnL a from Emericella nidulans NFCCI 3643. Trends Bioinform 2015;8:37-51. [CrossRef]
12. Hrmova M, Fincher GB. Structure-function relationships of b-D-glucan endo-and exohydrolases from higher plants. Plant Mol Biol 2001;47:73-91. [CrossRef]
13. George J, Arunachalam R, Paulkumar K, Wesely EG, Shiburaj S, Annadurai G. Characterization and phylogenetic analysis of cellulase producing Streptomyces noboritoensis SPKC1. Interdiscip Sci 2010;2:205-12. [CrossRef]
14. da Vinha FN, Gravina-Oliveira MP, Franco MN. Cellulase production by Streptomyces viridobrunneus SCPE-09 using lignocellulosic biomass as inducer substrate. Appl Biochem Biotechnol 2011;164:256-67. [CrossRef]
15. Murray MG, Thompson WF. Rapid isolation of high molecular weight plant DNA. Nucleic Acids Res 1980;8:4321-5. [CrossRef]
16. Hall TA. BioEdit: A user-friendly biological sequence alignment editor and analysis program for windows 95/98/NT. Nucleic Acids Symp Ser 1999;41:95-8.
17. Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol Biol Evol 2013;30:772-80. [CrossRef]
18. Robert X, Gouet P. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res 2014;42:W320-4. [CrossRef]
19. Gasteiger E, Hoogland C, Gattiker A. Protein identification and analysis tools on the ExPASy server. In: John M, editor. Walker, the Proteomics Protocols Handbook. United States: Humana Press; 2005. p. 571-607. [CrossRef]
20. Kumar S, Stecher G, Li M, Knyaz C, Tamura K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol 2018;35:1547-9. [CrossRef]
21. Marchler-Bauer A. CDD: Conserved domains and protein three-dimensional structure. Nucleic Acids Res 2013;41:384-52. [CrossRef]
22. Yin Y, Mao X, Yang J, Chen X, Mao F, Ying X. dbCAN: A web resource for automated carbohydrate-active enzyme annotation. Nucleic Acids Res 2012;40:445-51. [CrossRef]
23. McGuffin LJ, Bryson K, Jones DT. The PSIPRED protein structure prediction server. Bioinformatics 2000;16:404-5. [CrossRef]
24. Roy A, Kucukural A, Zhang Y. I-TASSER: A unified platform for automated protein structure and function prediction. Nat Protoc 2010;5:725-38. [CrossRef]
25. Halgren T. Identifying and characterizing binding sites and assessing druggability. J Chem Inf Model 2009;49:377-89. [CrossRef]
26. Friesner RA, Murphy RB, Repasky MP, Frye LL, Greenwood JR, Halgren TA, et al. Extra precision glide: Docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J Med Chem 2006;49:6177-96. [CrossRef]
27. Leo VV, Asem D, Zothanpuia, Singh BP. Actinobacteria: A highly potent source for holocellulose degrading enzymes. In: Singh JS, Singh DP, editors. Actinobacteria, New and Future Developments in Microbial Biotechnology and Bioengineering. Amsterdam: Elsevier; 2018. p. 191-205. [CrossRef]
28. Tomotsune K, Kasuga K, Tsuchida M, Shimura Y, Kobayashi M, Agematsu H, et al. Cloning and sequence analysis of the cellulase genes isolated from two cellulolytic streptomycetes and their heterologous expression in Streptomyces lividans. Int J Soc Mater Eng Resour 2014;20:213-8. [CrossRef]
29. Takasuka TE, Book AJ, Lewin GR, Currie CR, Fox BG. Aerobic deconstruction of cellulosic biomass by an insect associated Streptomyces. Science 2013;3:1-10. [CrossRef]
30. Lopez-Casado G, Urbanowicz BR, Damasceno CM, Rose JK. Plant glycosyl hydrolases and biofuels: A natural marriage. Curr Opin Plant Biol 2008;11:329-37. [CrossRef]
31. Hakkinen M, Arvas M, Oja M, Aro N, Penttila M, Saloheimo M, et al. Reannotation of the CAZy genes of Trichoderma reesei and transcription in the presence of lignocellulosic substrates. Microb Cell Fact 2012;11:134. [CrossRef]
32. Teegardin KA, James S, Barabote RD. Bioinformatic analysis of glycoside hydrolases in the proteomes of mesophilic and thermophilic Actinobacteria. MOJ Proteomics Bioinform 2017;5:75-81. [CrossRef]
33. Tomme P, Diane P, Emily DA, Robert AC, Miller JR, Antony R, et al. Comparison of a fungal (Family I) and bacterial (Family II) cellulose-binding domain. J Bacteriol 1995;117:4356-63. [CrossRef]
34. Jensen MS, Fredriksen L, MacKenzie AK, Pope PB, Leiros I, Chylenski P. Discovery and characterization of a thermostable two-domain GH6 endoglucanase from a compost metagenome. PLoS One 2018;13:e0197862. [CrossRef]
35. Sammond DW, Payne CM, Brunecky R, Himmel ME, Crowley MF, Gregg T, et al. Cellulase linkers are optimized based on domain type and function: Insights from sequence analysis, biophysical measurements, and molecular simulation. PLoS One 2012;7:e48615. [CrossRef]
36. Gilkes NR, Claeyssens M, Aebersold R, Henrissat B, Meinke A, Morrison HD, et al. Structural and functional relationships in two families of beta-1, 4-glycanases. Eur J Biochem 1991;202:367-77. [CrossRef]
37. Temuujina U, Chia W, Chang YB, Honga S. Identification and biochemical characterization of SCO3487 from Streptomyces coelicolor A3(2), an exo-and endo-type-agarase producing neoagarobiose. J Bacteriol 2012;194:142-9. [CrossRef]
38. Tanabe Y, Pang Z, Oda M. Cloning and sequencing of endo-1,3-b-glucanase from Cellulosimicrobium cellulans. J Biol Macromol 2008;8:60-3.
39. Juncosa M, Pons J, Dot T, Querol E, Planas A. Identification of active site carboxylic residues in Bacillus licheniformis 1,3-1,4-beta-D-glucan 4-glucanohydrolase by site-directed mutagenesis. J Biol Chem 1994;269:14530-5.
40. Michel G, Chantalat L, Duee E, Barbeyron T, Henrissat B, Kloareg B, et al. The kappa-carrageenase of P. carrageenovora features a tunnel-shaped active site: A novel insight in the evolution of clan-B glycoside hydrolases. Structure 2001;9:513-25. [CrossRef]
41. Fuchs KP, Zverlov VV, Velikodvorskaya GA, Lottspeich F, Schwarz WH. Lic16A of Clostridium thermocellum, a non-cellulosomal, highly complex endo-b-1,3-glucanase bound to the outer cell surface. Microbiology 2003;149:1021-31. [CrossRef]
42. Krah M, Misselwitz R, Politz O, Thomsen KK, Welfle H, Borriss R. The laminarinase from thermophilic eubacterium Rhodothermus marinus: Conformation, stability, and identification of active site carboxylic residues by site-directed mutagenesis. Eur J Biochem 2003;257:2101-11. [CrossRef]
43. Ize B, Stanley NR, Buchanan G, Palmer T. Role of the Escherichia coli Tat pathway in outer membrane integrity. Mol Microbiol 2003;48:1183-93. [CrossRef]
44. Anne J, Vrancken K, Mellaert LV, Impe JV, Bernaerts K. Protein secretion biotechnology in gram-positive bacteria with special emphasis on Streptomyces lividans. Biochim Biophys Acta 2014;1843:1750-61. [CrossRef]
45. Abraham A, Narayanan SP, Philip S, Nair DG, Chandrasekharan A, Kochupurackal J. In silico characterization of a novel b-1,3-glucanase gene from Bacillus amyloliquefaciens-a bacterial endophyte of Hevea brasiliensis antagonistic to Phytophthora meadii. J Mol Model 2013;19:999-1007. [CrossRef]
46. Sahay A, Shakya M. In silico analysis and homology modelling of antioxidant proteins of spinach. J Proteomics Bioinform 2010;3:148-54. [CrossRef]
47. Ikai A. Thermostability and aliphatic index of globular proteins. J Biochem 1980;88:1895-8.
48. Deshmukh RA, Jagtap S, Mandal MK, Mandal SK. Purification, biochemical characterization and structural modelling of alkali-stable b-1,4-xylan xylanohydrolase from Aspergillus fumigatus R1 isolated from soil. BMC Biotechnol 2016;16:11. [CrossRef]
49. Sandgren M, Wu M, Karkehabadi S, Mitchinson C, Kelemen BR, Larenas EA, et al. The structure of a bacterial cellobiohydrolase: The catalytic core of the Thermobifida fusca family GH6 cellobiohydrolase Cel6B. J Mol Biol 2013;425:622-35. [CrossRef]
50. Hong TY, Hsiao YY, Meng M, Li TT. The 1.5 a structure of endo-1, 3-beta-glucanase from Streptomyces sioyaensis: Evolution of the active-site structure for 1, 3-beta-glucan-binding specificity and hydrolysis. Acta Crystallogr D 2008;64:964-70. [CrossRef]
51. Wu M, Nerinckx W, Piens K, Ishida T, Hansson H, Sandgren M, et al. Rational design, synthesis, evaluation and enzyme-substrate structures of improved fluorogenic substrates for family 6 glycoside hydrolases. FEBS J 2013;80:184-98. [CrossRef]
52. Chinnathambi V, Balasubramanium M, Gurusamy R, Paramasamy G. Molecular cloning and expression of a family 6 cellobiohydrolase gene cbhII from Penicillium funiculosum NCL1. Adv Biosci Biotechnol 2015;6:213-22. [CrossRef]
Supplementary Material
 | Supplementary Figure 1: Cloning of ENDO13 and EXO14 genes in pGEM®-T Easy Vector (a) isolated Genomic DNA, Lane 1: Streptomyces althioticus TBG-MR17, Lane 2: Streptomyces cinereoruber subsp. cinereoruber TBG-AL13 (b) the PCR amplicons of ENDO13 (~1400 bp) and EXO14 (~2000 bp) genes (c) construction of pGEMT/EXO14 and pGEMT/ENDO13 plasmid (d) screening of positive colonies by colony PCR. Lane 1-5: Positive colonies with ENDO13 gene, Lane M: 1 kb DNA ladder, Lane 6-10: Positive colonies with EXO14 gene (e) confirmation of the presence of gene of interest in isolated plasmids. Lane 1: PCR amplification of pGEMT/ENDO13 with gene specific primers, Lane 2: PCR amplification of pGEMT/EXO14 with gene specific primers, Lane 3: pGEMT/ENDO13 plasmid, Lane 4: pGEMT/EXO13 plasmid, Lane 5: PCR amplification of pGEMT/ENDO13 with Tvect primers, Lane 6: PCR amplification of pGEMT/EXO14 with Tvect primers
[Click here to view] |
 | Supplementary Figure 2: Aligned gene sequences obtained after sequencing and frame analysis. (a) EXO-14 sequence, 5’3’frame 1 showed 1737 bp full gene sequence. (b) ENDO-13 sequence, 5’3’frame 3 showed 1293 bp full gene sequence. Yellow color sequence denotes the pGEMT vector sequences, green, and red codons represents start and stop codons, respectively
[Click here to view] |
 | Supplementary Figure 3: The ORF of EXO-14 gene with corresponding putative amino acids
[Click here to view] |
.png) | Supplementary Figure 4: The ORF of ENDO-13 gene with corresponding putative amino acids
[Click here to view] |
 | Supplementary Figure 5: Conserved domain search of EXO-14 protein. (a) NCBI conserved domain search based on sequence alignment recognized EXO-14 has linker which separates N-terminal CBM2 family and C-terminal GH6 family domains. (b) dbCAN domain search also confirmed the above domain architecture and the protein belongs to GH6 CAZyme family
[Click here to view] |
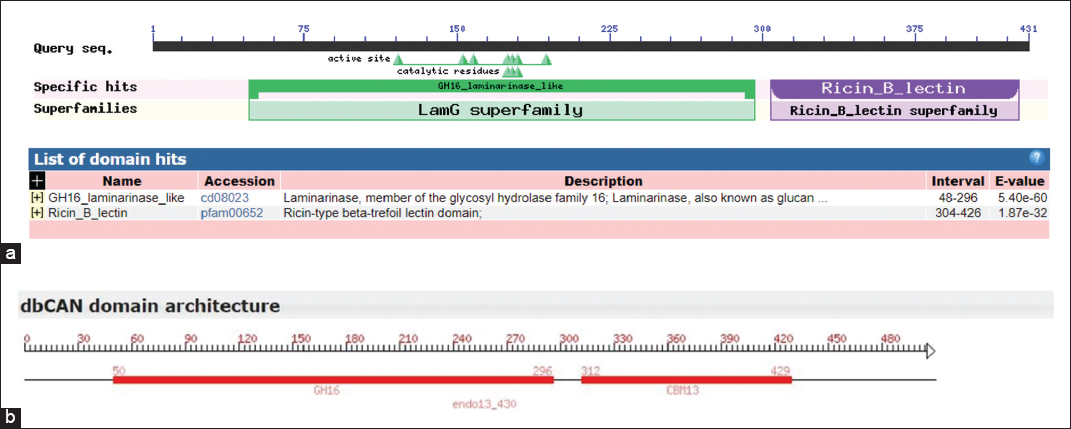 | Supplementary Figure 6: Conserved domain search of ENDO13 protein. (a) NCBI conserved domain search based on sequence alignment recognized ENDO-13 has N-terminal GH16-laminarinase-like domain and C-terminal Ricin-B-lectin family domains, both are separated by a linker sequence. (b) dbCAN domain architecture of ENDO-13 confirmed, the protein belongs to GH16 CAZyme family and Ricin-B-lectin domain was a carbohydrate binding module 13 (CBM13) family of CAZy database
[Click here to view] |
 | Supplementary Figure 7: Signal peptide prediction of EXO-14 and ENDO-13 proteins. (a) Predicted signal sequence of EXO-14 protein showed cleavage site in between A-32 and A-33 with cutoff value 0.450. (b) Predicted signal sequence of EXO-14 protein showed a cleavage site in between A-35 and A-36 residues with 0.450 cutoff
[Click here to view] |

.png)
.png)
.png)
.png)
.png)
.png)