
1. INTRODUCTION
Vibriosis is a typical systemic bacterial type of infection. It is caused by bacteria belonging to the family Vibrionaceae. Vibriosis is often considered a significant problem worldwide in the aquaculture industry. The common factors behind the cause of vibriosis in fishes are fluctuation of physicochemical properties of water as well as overpopulation. Previously, vibriosis has been responsible for causing significant economic loss in the shrimp aquaculture industry. Recently, vibriosis has also been responsible for causing losses associated with various species of cage-cultured fishes worldwide [1,2].
The vibrio class of bacteria has been reported to interfere with the mode of action of various drugs, especially antibiotics. They act by modifying the functional group of the antibiotics, thereby rendering them ineffective [3,4]. Chloramphenicol acetyltransferase is one such antibiotic resistance enzyme present in the vibrio class of bacteria. This particular enzyme of the bacteria provides resistance against chloramphenicol type of drugs by acetylating the C-3 hydroxyl group of the drug, thereby preventing them from binding to the 50S subunit of the bacterial ribosome. Thus, the bacterial protein translation process is not stopped [5,6].
Florfenicol is one such drug that has been specifically used to treat vibriosis worldwide. Florfenicol antibiotic primarily provides bacteriostatic action and has been reported to show similar activity as that of chloramphenicol [7,8]. Previous researches have stated that florfenicol functions by binding to the bacterial ribosomal subunits leading to inhibition of the bacterial acetyltransferase enzyme. Moreover, the bacterial receptor target of action of florfenicol has been found to be the same as that of thiamphenicol and chloramphenicol. Furthermore, the structure of florfenicol contains fluorine atoms in the place of hydroxyl group at C-3 position in the structure of thiamphenicol and chloramphenicol. The presence of the fluorine group helps florfenicol to be less susceptible to bacterial deactivation, thereby making it more effective than chloramphenicol and thiamphenicol. In spite of all this positivity, florfenicol has been found to show poor drug properties as well as adverse effects [9,10].
The main aim to be achieved through this study is to design new analogs of florfenicol that can target bacterial acetyltransferase and exhibit better drug properties as well as fewer side effects. Using florfenicol as the base framework, the primary and secondary modifications were done by using Chemsketch software. All these modified molecules were then screened against all the drug-likeness rules. Following this, ADMET analysis of the modified molecules is also performed. After these steps, the configured molecules were docked in the active sites of the bacterial acetyltransferase enzyme. The results obtained from the study showed that the designed molecules can be considered as potential lead molecules in inhibiting bacterial chloramphenicol acetyltransferase enzymes, which could ultimately be used to treat vibriosis [11,12].
2. MATERIALS AND METHODS
2.1. Molecular Target and Active Site Determination
The bacterial chloramphenicol acetyltransferase is the main target which is responsible for providing resistance against antibiotics administered in vibriosis. The molecular target structure was retrieved from the Protein Data Bank (PDB). The name of the PDB-ID retrieved for the study is 6PXA. The protein 6PXA contains preexisting native ligand chloramphenicol that shows the active pockets. The unnecessary native ligands and water molecules were first cleaned using PyMOL software [13,14]. The binding pockets present on the protein were visualized using PyMOL & Discovery Studio, based on the co-crystallized ligands and also the inbuilt AutoDock Vina protocol that predicts the binding site during protein–ligand docking process [15,16].
2.2. Docking of Florfenicol with Chloramphenicol Acetyltransferase
The Florfenicol molecule was used as the basic framework for designing effective lead molecules. The structure of Florfenicol was retrieved from the pubchem database in the form of SDF format. Ligand structure was optimized using “uff” forcefield and conjugate gradients algorithm, available within PyRx-Open Babel tool. This structure was then docked with the molecular target chloramphenicol acetyltransferase. The binding affinity obtained from docking is then used as the standard reference for further modified structures. The docking study was done using PyRx (Autodock Vina), computational drug discovery and virtual screening software used for screening libraries of compounds against potential drug targets [14,17].
2.3. Initial Ligand Modifications Using Florfenicol Framework
Different molecules were designed using florfenicol as the basic framework for modification. The molecules were drawn and designed using Chemsketch software. The newly sketched molecules were then subjected to various in silico analysis. Basically, to fit perfectly into the active pocket of the protein, initial lead modifications were performed.
2.4. Primary Lead Development and Drug Screening
The OSIRIS property explorer tool was used to modify the lead molecule. The structure of florfenicol was assessed in the stand-alone Java Runtime Environment. It was then modified by substitution or deletion or by the addition of atoms in a manner such that the skeleton structure of the lead molecule was retained. The modifications were also done to increase both solubility and polarity as well as reducing unnecessary side effects. Using Lipinski’s rule of five, ADMET analysis was done using physiological parameters such as LogP, solubility, topological polar surface area (TPSA), molecular weight, drug likeness, and drug score. All the values were noted. The color code indicated the severity of the unnecessary side effect. The green color indicated no unnecessary side effects. The red color indicated severe side effects. Orange and yellow colors denoted medium and low risk of unnecessary side effects respectively. The better properties showing designed molecules were then draw in Chemsketch. Following this, the molecules were saved in a PDB format for further docking analysis.
2.5. Docking and Selection of the Lead Molecule
The docking procedure was done using PyRx (Autodock Vina). The proteins were prepared first using Autodock tools. After this step, the protein was introduced into PyRx. The modified molecules were then introduced into PyRx. The energy minimization of the molecules was done using “uff” forcefield and conjugate gradients algorithm, available within PyRx-Open Babel tool. Following this, docking was performed. The results were displayed in terms of binding affinity. The binding affinity represents the binding energy. The binding energy exhibits the extent of binding of a particular molecule. Furthermore, the best type of configuration would be the one that would bind with its target. The binding affinity of chloramphenicol acetyltransferase with the initially modified ligands should be more than the binding affinity of florfenicol with chloramphenicol acetyltransferase. The molecule with the best binding affinity and good ADME property was chosen for further analysis.
2.6. Secondary Ligand Modifications of Initial Ligand
Further alterations were made to design new lead molecules using the best initial ligand modification. The secondary lead modifications were aimed for increasing stability. This was mainly done for acquiring better binding affinity between new lead molecules with chloramphenicol acetyltransferase. The structures were drawn, modified, and designed using Chemsketch. Following this, the molecules were subjected to in silico analysis similarly as that of the initial ligand modifications.
2.7. Secondary Lead Development and Drug Screening
The secondary ligand modifications were also designed using OSIRIS property explorer tool. The designed molecules were then assessed in the stand-alone Java Runtime Environment. The molecules were designed in such a manner so that the skeletal structure of the lead molecule was retained. Furthermore, modifications were aimed at improving polarity and solubility as well as reducing unnecessary side effects. Using Lipinski’s rule of five, the ADME analysis of the secondary lead molecules was performed. Physicochemical parameters such as LogP, solubility, TPSA, molecular weight drug-likeness, and drug score were all noted down. The color code indicated the severity of the unnecessary side effect. The green color indicated no unnecessary side effects. The red color indicated severe side effects. Orange and yellow colors denoted medium and low risk of unnecessary side effects, respectively. The better properties showing designed molecules were then drawn in Chemsketch. Following this, the molecules were saved in the PDB format for further docking analysis.
2.8. Docking and Selection of Lead Molecule
The docking study was carried out using PyRx (Autodock Vina). The secondary ligand modifications were docked with the chloramphenicol acetyltransferase receptor. The binding affinity obtained from docking should be higher than that binding affinity of the initial lead modifications and the chloramphenicol acetyltransferase receptor. The lead molecules were selected based on higher binding affinity and better ADME properties than primary ligand modifications. More drug properties of the best lead molecule were further retrieved from Pre-ADMET web-based application [18]. Furthermore, structural analysis of the final lead molecule was also performed with the help of the discovery studio visualizer and PyMOL software.
3. RESULTS
3.1. Active Site of Chloramphenicol Acetyltransferase
Discovery Studio was used to identify, visualize, and analyze active inhibition sites for chloramphenicol acetyltransferase. For visualizing and identifying the structure of the target, PDB ID 6PXA was retrieved. A two-dimensional structure was then generated containing the receptor and its bound inhibitor. This was then used to identify the bound amino acid residues in the active site of the receptor. Identification of the bound residues was helpful in designing the ligands in such a manner so that they could easily fit into the active pocket of the target. The active site residues of 6PXA PDB identified were Phe 105, Phe 98, Ser 101, Glu 104, Phe 100, Pro 99, Gln 88, Ala 85, Leu 48, Phe 62, Phe 113, Gly 89. The gridbox for protein–ligand was set to encase all these active site residues.
3.2. Florfenicol Docking
A protein–ligand docking value was obtained between florfenicol and the chloramphenicol acetyltransferase receptor using PyRx (Autodock Vina). This docking score was then used as a base score for further design of the lead molecules. The binding energy obtained for florfenicol was −6.0 kcal/mol.
3.3. Primary Ligand Modifications of Florfenicol
Chemsketch was used to design the primary lead modifications. A total of 15 modifications was developed by altering the R groups present in the basic framework. These 15 modifications are displayed in Figure 1 and the modifications are summarized in Table 1. Primary modifications were performed to cover the active site pocket of the receptor effectively compared to the basic framework of the lead molecule. The active site of the protein is highlighted in a light green color, as compared to the allosteric portions of the protein.
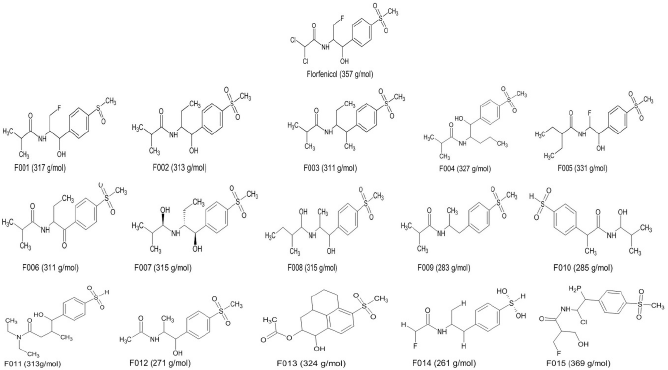 | Figure 1: Initial ligand modifications were done using Chemsketch, 15 structures were developed based on modifications. [Click here to view] |
 | Table 1: List of all initially modified ligands. [Click here to view] |
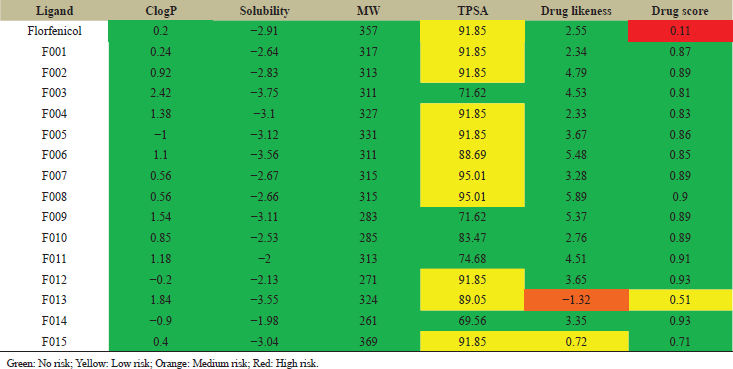 | Table 2: Lipinski’s rule of five analysis of florfenicol and primary modified ligands for ADMET properties. [Click here to view] |
3.4. Primary Lead Development and Drug Screening
The OSIRIS property explorer tool was used to modify the lead molecule florfenicol. The structure of florfenicol was assessed in the stand-alone Java Runtime Environment. The 15 modified ligands were then designed by substitution or deletion or by addition of atoms in a manner such that the skeleton structure of the lead molecule was retained. The 15 modifications were done basically to increase both solubility and polarity as well as reducing unnecessary side effects and toxicity (Table 2). Using Lipinski’s rule of five, ADMET analysis of the 15 modifications was done using physiological parameters such as LogP, solubility, TPSA, molecular weight, drug-likeness, and drug score. All the values were noted. The color code indicated the severity of the unnecessary side effects. The green color indicated no unnecessary side effects. The red color indicated severe side effects. Orange and yellow color denoted medium and low risk of unnecessary side effects respectively. The better property showing designed molecules was then draw in Chemsketch. Following this, the molecules were saved in the PDB format for further docking analysis. All 15 initially modified structures showed better ADME properties than the basic framework florfenicol. Moreover, all the primary modifications, when assessed on OSIRIS property explorer showed zero risks related to mutagenic, tumorigenic, irritant, and reproductive effect factors.
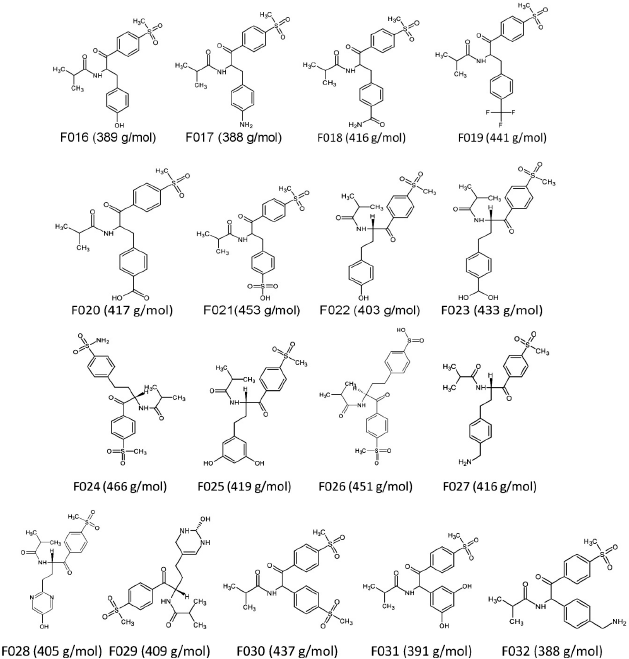 | Figure 2: F006 was taken as the skeletal structure to design 17 secondary structures. [Click here to view] |
3.5. Docking Results and Selection of Lead Molecule
The 15 modifications were now docked with the target chloramphenicol acetyltransferase (Table 3). The docking was performed on PyRx. Out of the 15 modifications, F002, F003, F004, F005, F006, F009, F010, F011, F012, F013, F014, and F015 showed docking scores of −6.5, −6.5, −6.8, −7.1, −7.3, −6.4, −6.7, −6.6, −6.8, −6.9, −6.2 and −6.3 kcal/mol respectively. Out of the 15 initially modified ligands, 12 of the ligands showed higher binding energy than the basic framework. From these 12 ligands, the further selection was made on the basis of higher binding affinity along with better ADME properties. Among these 12 selected ligands, F006 was chosen was for further secondary ligand development as it had the best docking score as well as good ADME properties. Furthermore, F006, when assessed on the Pre-ADMET web server, showed poor blood–brain barrier property with a value of 0.01222. It also displayed good water solubility property with a value of 19.318 as well as good plasma protein binding property with a value of 77.7702. Moreover, it also displayed poor skin permeability property with a value of −1.2802.
3.6. Secondary Ligand Modifications Using F006
Using F006 as the framework, secondary modifications were done. The modifications were done using Chemsketch. Better structures were developed by altering R-groups and by stabilizing hydrophilic hydrophobic interactions. 17 secondary modifications were developed from F006 and are displayed in Figure 2 and the chances are listed in Table 4.
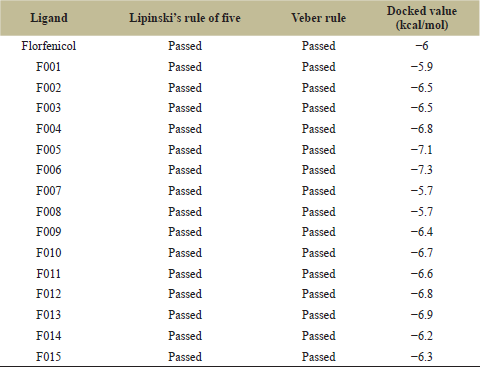 | Table 3: Drug screening and docking results for florfenicol and its primary modified ligands. [Click here to view] |
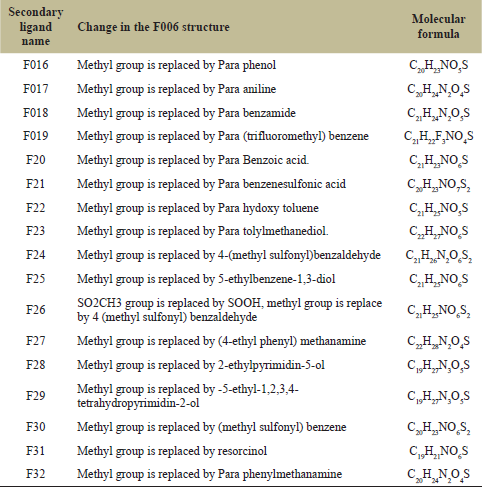 | Table 4: List of all secondary modified ligands. [Click here to view] |
3.7. Secondary Lead Development and Drug Screening
The OSIRIS property explorer tool was used to modify the lead molecule F006. The structure of florfenicol was assessed in the stand-alone Java Runtime Environment. The 17 secondary modified ligands were then designed by substitution or by the addition of atoms in a manner such that the skeleton structure of the lead molecule was retained (Table 5). The 17 modifications were done basically to increase solubility, polarity, binding affinity as well as reducing unnecessary side effects. Using Lipinski’s rule of five, ADMET analysis of the 17 secondary modifications was done using physiological parameters such as LogP, solubility, TPSA, molecular weight, drug-likeness, and drug score. All the values were noted. The color code indicated the severity of the unnecessary side effect. The green color indicated no unnecessary side effects. The red color indicated severe side effects. Orange and yellow colors denoted medium and low risk of unnecessary side effects respectively. The better properties showing designed molecules were then drawn in Chemsketch. Following this, the molecules were saved in PDB format for further docking analysis. All secondary modifications, when assessed on OSIRIS property explorer showed zero risks related to mutagenic, tumorigenic, irritant, and reproductive effect factors except F021, which showed a high risk of mutagenicity.
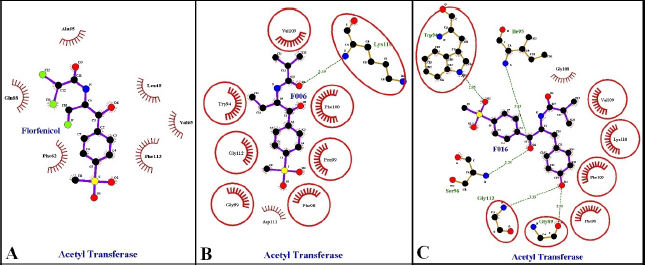 | Figure 3: This figure compares 2D amino acid interactions of F016, F06 and florfenicol with the receptor bacterial chloramphenicol acetyltransferase generated with the help of Ligplus application. A represents amino acid interactions of florfenicol, B represents amino acid interactions of F06, C represents amino acid interactions of F016. [Click here to view] |
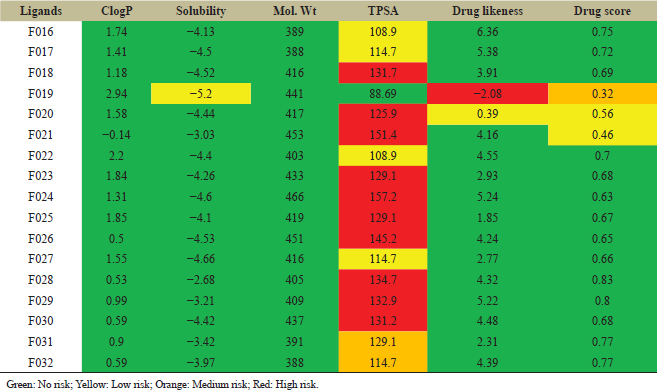 | Table 5: Lipinski’s rule of five analysis of secondary modified ligands for ADMET properties. [Click here to view] |
3.8. Docking Results and Selection of Lead Molecule
The 17 new secondary modified ligands were now docked with chloramphenicol acetyltransferase (Table 6). All the secondary modified ligands showed better docking scores than the basic framework F006. But among these F016 was chosen as it showed a better docking score of −8.6 kcal/mol along with good ADME properties. More drug properties of F016 were further retrieved from Pre-ADMET web-based application. F016, when assessed on the Pre-ADMET web server showed poor blood–brain Barrier property with a value of 0.02242. It also has good plasma protein binding property with a value of 86.2747, good water solubility property with a value of 10.4255, and poor skin permeability property with a value of −1.6579. Structural analysis of F016 was also performed using ligplus and PyMOL software.
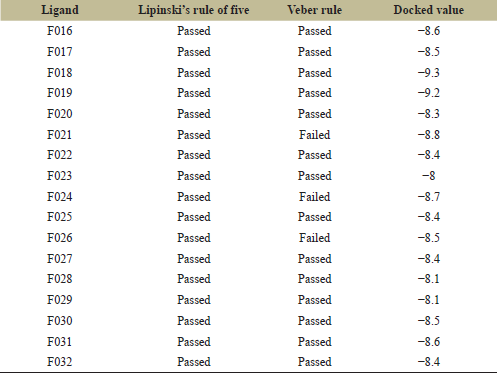 | Table 6: Drug screening and docking results of secondary modified ligands. [Click here to view] |
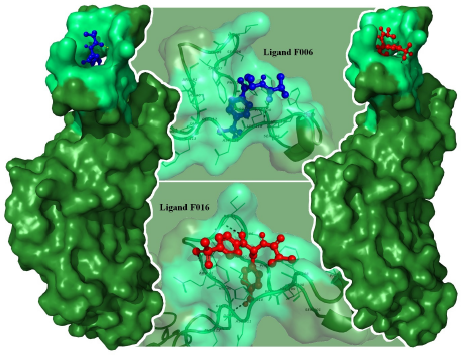 | Figure 4: PyMOL visualization of chloramphenicol acetyltransferase with F016 (red) and F06 (blue). The apoprotein is represented by dark green color. The active sites are represented by light green color. [Click here to view] |
4. DISCUSSION
Vibriosis has been considered a major problem worldwide in the aquaculture industry. Moreover, there has not been much qualitative research on vibriosis or organisms responsible for causing vibriosis. With the help of this study, we were able to design a molecule for targeting chloramphenicol acetyltransferase, a major enzyme present in vibriosis-causing bacteria, which is responsible for providing resistance against antibiotics.
With the help of the literature survey, we have found out studies that have clearly stated bacterial chloramphenicol acetyltransferase’s importance in vibrio species. The literature survey has also helped us to isolate our main basic framework, florfenicol. The basic framework of florfenicol was modified to produce 15 primary structures and 17 secondary structures. These modified structures were then subjected to docking, screening, and ADME analysis. We were able to design a molecule, F016 with the help of this study which has shown good binding affinity with bacterial chloramphenicol acetyltransferase receptor along with good ADME properties (Figure 3 and Figure 4). The F016 also showed similar interactions with bacterial acetyltransferase like florfenicol, thereby making it an ideal molecule to inhibit chloramphenicol acetyltransferase. Lastly, we can conclude by saying that F016 can be considered as a lead candidate in binding with chloramphenicol acetyltransferase, and hence can be used for treating vibriosis.
5. CONCLUSION
The results of this study conclude that the drug target chloramphenicol acetyltransferase protein can be used for developing antibiotics against vibriosis infection in aquaculture management. Florfenicol structure was taken as a skeleton and was modified to develop 15 ligands that were screened against the drug target. F006 was used as skeleton structure for secondary modification. Among all the developed modifications of the ligand, the ligand molecule F016 ligand was identified as the best modified ligand that shows greater inhibition potential against chloramphenicol acetyltransferase protein and demonstrates significantly better ADME property. This molecule should further be investigated in-vitro to justify the given hypothesis.
6. ACKNOWLEDGMENTS
The authors thank SciWris Life Sciences for supporting this study.
7. CONFLICT OF INTERSET
No known conflict of interest for this study.
8. FUNDING
No funding was availed for this study.
9. ETHICAL APPROVALS
This study does not involve experiments on animals or human subjects.
REFERENCES
1. Mohamad N, Amal MNA, Yasin ISM, Saad MZ, Nasruddin NS, Al-saari N, et al. Vibriosis in cultured marine fishes: a review. Aquaculture 2019;512(May):1–17. CrossRef
2. Dunn AK. Vibrio fischeri metabolism: symbiosis and beyond [Internet]. 1st edition. In: Advances in Microbial Physiology, Elsevier Ltd, Amsterdam, Netherlands, pp 37–68, vol 61, 2012. CrossRef
3. Seljestokken B, Bergh, Melingen GO, Rudra H, Hetlelid Olsen R, Samuelsen OB. Treating experimentally induced vibriosis (Listonella anguillarum) in cod, Gadus morhua L., with florfenicol. J Fish Dis 2006;29(12):737–42. CrossRef
4. Samuelsen OB, Bergh Ø. Efficacy of orally administered florfenicol and oxolinic acid for the treatment of vibriosis in cod (Gadus morhua). Aquaculture 2004;235(1–4):27–35. CrossRef
5. Wang L, Han YN, Jin S, Ma Y, Wang GL, Zhao QS, et al. Pharmacokinetic study of florfenicol in healthy and vibriosis-infected pseudosciaena crocea after oral administration. J Appl Biol Chem 2015;58(4):363–8. CrossRef
6. Parmar P, Yusufzai SKI, Parmar H V, Nanjiyani RP, Chavda VM. Therapeutic potentiality of florfenicol against vibriosis in Litopenaeus vannamei therapeutic potentiality of florfenicol against vibriosis in Litopenaeus vannamei. J Entomol Zool Stud 2018;6(August):463–7.
7. Jangaran Nejad A, Peyghan R, Najafzadeh Varzi H, Shahriyari A. Florfenicol pharmacokinetics following intravenous and oral administrations and its elimination after oral and bath administrations in common carp (Cyprinus carpio). Vet Res forum an Int Q J 2017;8(4):327–31.
8. Convention USP. FLORFENICOL (Veterinary—Systemic) pharmacology/pharmacokinetics. United States Pharmacopeial Convention, North Bethesda, MD, pp 1–6, 2007.
9. Papich MG DVM, MS, DACVCP. Florfenicol. In: Saunders Handbook Veterinary Drugs, St.Louis, MO: Saunders Elsevier, pp 327–9, 2016. CrossRef
10. Martos P, Shurmer B. Sample preparation techniques for the determination of veterinary drugs in food matrices [Internet]. In: Comprehensive Sampling and Sample Preparation, Elsevier, Amsterdam, Netherlands, pp 405–14, vol 4, 2012. CrossRef
11. Sreenivas A, Menta V, Ravi L. Designing of ligand-107 an effective variant of antimalarial drug lumefantrine through structure-based computer-aided drug development approach. Asian. J Pharm 2020;14(36):2–9.
12. Sadeesh N, Camil Rex M, Ravi L. Structure-based drug designing for n-methyl-d-aspartate receptor: link between neurodegenerative disease and glioblastoma. J Appl Biol Biotechnol 2021;9(1):25–31.
13. DeLano WL. The PyMOL molecular graphics system, version 2.3. Schrödinger LLC, New York, NY, 2020.
14. Seeliger D, De Groot BL. Ligand docking and binding site analysis with PyMOL and Autodock/Vina. J Comput Aided Mol Des 2010;24(5):417–22. CrossRef
15. Visualizer DS. v4. 0.100. 13345. Accelrys Software Inc, San Diego, CA, 2005.
16. Studio D. Dassault systemes BIOVIA, siscovery studio modelling environment, release 4.5. Accelrys Software Inc, San Diego, CA, 2015
17. Dallakyan S, Olson AJ. Small-molecule library screening by docking with PyRx. Methods Mol Biol 2015;1263:243–50. CrossRef
18. Lee SK, Lee IH, Kim HJ, Chang GS, Chung JE, No KT. The PreADME approach: web-based program for rapid prediction of physico-chemical, drug absorption and drug-like properties. EuroQSAR 2002 Des Drugs Crop Prot Process Probl Solut [Internet] 2002;418–20. Available via https://preadmet.bmdrc.kr/toxicity-prediction/