
1. INTRODUCTION
Arachis hypogaea L. (Groundnut) is the most important legume and edible oil seed crop grown worldwide. Fusarium oxysporum that causes vascular wilt, root, and crown rots diseases is causing damage in economically imperative crops A. hypogaea L. [1]. F. oxysporum consisting of 70 forme speciales, it can cause diseases on a variety of plant species and has recently been recognized as the fifth most important pathogenic plant fungus [2]. Earlier scientists has applied RNA-seq technology successfully onto species like humans, yeast, and other important agricultural crops [3-5]. Advanced studies of F. oxysporum f. sp. lycopersici (Fol) genome in tomato, reveals that genes infecting strain F. oxysporum lycopersici (Fol) which to understand the role genes of F. oxysporum. [6]. This study led to the identification of specific pathogenicity chromosomes and virulence factors in genome of F. oxysporum. Secondary metabolites (SM) are organic molecules of low-molecular-weight with diverse chemical structures and biological activities and are produced to confer a selective advantage to the organism. Even though Fusarium SM is crucial for disease development, the major metabolites in the scientific study of Fusarium not explained.
Fusaric acid (FA) (5-butylpicolinic acid) derived SM produced by Fusarium species including F. oxysporum members of Fusarium fujikuroi species complex (FFSC) [7]. FA was first discovered in laboratory culture Fusarium heterosporum Nees [8] and a phytotoxin implicated in the pathogenesis of Fusarium wilts [9]. It is still renowned for its strong phytotoxicity [10] and to chelate metal ions inside tomato [11]. While many investigations have been carried out to determine the size of FA biological activity in pathogenesis, the mechanism of genes and its toxic effects are still remained to be elucidated.
There are no studies related to FA toxicity in A. hypogaea L. Among the toxic metabolites, secreted by F. oxysporum FA was confirmed by HPLC-MS/MS method in A. hypogaea L. [12]. FA biosynthetic gene (FUB) cluster was first identified in two FFSC species, namely, Fusarium verticillioides, the causal fungus of stalk rot of maize [13], and F. fujikuroi, the causal fungus of bakanae disease in rice [14].
Earlier workers confirmed that Fusarium FUB cluster consisting of 12 genes (FUB1 to FUB12) [15,16]. The functions of the FUB genes in Fusarium were predicted through the functions of their annotated homologs, including a polyketide synthase (PKS, FUB1), a protein (FUB1), eight different enzymes (FUB3-FUB8), two transcription factors (TFs) (FUB10 and FUB12), and a transporter (FUB12). Prior reports affirmed that the genes involved in the biosynthesis of similar SM are co-coordinately managed and located adjacent to one other in the genome [17,18]. These biosynthetic gene clusters usually include a key gene encoding an enzyme PKS that catalyzes assembly of the SM chemical scaffold and a majority of cluster genes, at least one TF that regulates gene expression within the cluster and a protein that transports the SM out of the cell. The studies of FUB gene cluster in different species were identified but its role in A. hypogaea L. and its mechanism of genes in infection is still lacking.
Transcriptomic information is used in a wide range of biological studies and provides fundamental insights into biological processes and applications. Next-generation sequencing technologies have advanced rapidly with ribonucleic acid sequencing (RNA-seq) and becoming an instrumental assay for the analysis of fungal transcriptomes, particularly in the levels of gene expression [19], the gene expression profiles during development [5,20] or gene discovery; however, transcriptomic information provides a little information for the peanut plant and no scientific report is available regarding the FUB genes of F. oxysporum infecting A. hypogaea L. However, first report enlightening symbiosis-related genes in a genome-wide manner in peanut [21].
As studies on Fusarium-Arachis pathos-system are only limited to initial stages of infection and wilt control, molecular information about Fusarium-Arachis interaction is not available. There are no studies that deal with the expression analysis of pathogenicity-related genes in Fusarium-Arachis pathos-system. The manuscript deals with the transcriptomic profile of Fusarium infected Arachis plants which provide insight into the genes that are responsible for virulence pathogenicity of Fusarium and elucidating the role of these genes in infection progression, provide insight into genetic pathways required for FA biosynthesis. Workers demonstrated that Trichoderma spp. may inhibit FA production in roots and leaves of peanut or maize infected with Fusarium spp. [22,23]. The efficient role of 6PAP in the inhibition of biosynthesis of FA and its degradation has been described in Pseudomonas fluorescens [24]. Earlier studies have proved that the combined application of Trichoderma with Pseudomonas strains has improved activity than individual strains in biological control of diseases [25,26].
The present study concentrates on identification of FUB genes responsible for FA production secreted by the F. oxysporum causing Fusarium wilt in A. hypogaea and specific interactions of combination of biocontrol agents (Trichoderma viride and P. fluorescens) with the pathogen on the expression of FUB genes indicating the potential benefits of combination of biocontrol agents can be used for the inhibition of FA and reducing the toxicity of FA in A. hypogaea L.
2. MATERIALS AND METHODS
2.1. Plant Material
Plants of A. hypogaea L. raised from the seeds (JLR-variety) were grown in each of nine earthen pots (25 cm diameter) up to 75 DAS and categorized into three sets. Control plants – first set of three pots was sprayed with distilled water (sample 1). Infected plants – second set of three pots were sprayed with a conidial suspension of F. oxysporum (MTCC 2087), that is, 1×108 spores / mL on 30 DAS and left without any treatment (sample 2). Infected treated plants – third set of pots three pots were sprayed with pathogen on 30 DAS. F. oxysporum infected plants were sprayed with OIC of culture filtrates of combinations of biocontrol agents, T. viride (MTCC 2047) + P. fluorescens (MTCC644) (Tv 1% + Pf 2%) on 40 DAS as sample 3. The optimum inhibitory concentration of different biocontrol agents was determined based on the results of conidial germination and mycelial growth. On 50 DAS, the leaves of control, infected, infected treated plants collected, and used for RNA sequencing.
2.2. Transcriptome Profiling
Three samples of control, infected (inoculated with F. oxysporum), and treated plants leaves from 50DAS were collected. All samples were flash frozen in liquid nitrogen and stored at -800C until needed. Nucleic acid isolation was carried out based on a protocol ready-to-use TRIzol® Reagent (Invitrogen, USA) following the manufacturer’s instructions. Each sample was maintained in three biological replicates and RNA was extracted from each of three replicates. Polyadenylated (Poly(A)) mRNA was isolated from the total RNA, and then cDNA libraries were synthesized and sequenced at the 101bp paired-end read mode with Illumina HiSeq®4000.
The leaf samples of control, infected, and treated were aligned to A. hypogaea L. genome from NCBI. Read count for aligned reads was fetched using tool feature count. High-quality reads (fast format) were aligned to A. hypogaea genome (from NCBI) using TopHat splice aware aligner. TopHat is an advanced fast splice junction used for mapper in RNA-Seq reads. It aligns RNA-Seq reads to genomes using the ultra-high-throughput short read aligner Bowtie2 [27].
Differential expression analysis was performed as “Treated versus Control” and “Infected versus Control” (Host-Specific Expression). The transcripts with log2 fold Change ≥2 and P ≤ 0.05 were considered as significantly upregulated and the transcripts with log2 fold Change ≤−2 were and P ≤ 0.05 considered as significantly downregulated. Sample specific transcripts were identified as well.
Unaligned reads of infected and treated samples were aligned to F. oxysporum genome from NCBI using Bowtie2. Read count for aligned reads that aligned to Fusarium was fetched software using feature count.
2.3. Differential Expression Analysis w.r.t (Pathogen-Specific Expression)
Reads of infected and treated sample that did not align to A. hypogaea L. were aligned with F. oxysporum genome from NCBI using Bowtie2 aligner. Later the read counts of these aligned reads were taken using tool feature count. These read counts were taken for differential expression analysis (Pathogen-Specific Expression).
Differential expression analysis was performed using DESeq R package. Differential expression analysis was done as “treated vs. infected.” The transcripts with log2 fold Change values are taken as upregulated/downregulated without any P-value significance. Hence, these transcripts are not statistically significant. There are very few transcripts expressed commonly in both the treated and infected sample at same time, so it is showing very less differentials. However, there are sample specific transcripts.
2.4. Gene Ontology and Pathways
Gene ontology and pathways were found by blastx hitting infected and treated sequences against Uniprot_Fungus sequences, and top five GO and pathways w.r.t pathogen for treated versus infected differential genes is depicted.
2.5. cDNA Synthesis and Gene Expression Analysis and Amplification by Real-time Polymerase Chain Reaction (RT-PCR)
The samples control, treated, and infected were analyzed for FUB1 and FUB11 genes expression. From the test samples, total RNA was extracted using RNA isolation reagent (TRIzol method). From the extracted Total RNA, 1000 ng was used for the conversion of cDNA synthesis and cDNA was synthesized (reverse transcription). qPCR amplification was carried out using gene specific primers. The RNA and Oligo dT/Random hexamer primers were used for the first strand cDNA synthesis by reverse transcriptase. The RT-PCR reaction volume of 20 μl containing 2 μl of cDNA and 10 μl of SYBR Green Super mix (Bio Rad, USA) under the following conditions denaturation at 95°C for 30 sec, annealing at 55°C (Gradient) for 30 sec, extension of 35 cycles at 95°C for 30 sec in Bio-Rad CFX96 system. The mRNA expression levels were normalized to that of housekeeping gene and the results were analyzed. The genes under study were first standardized and amplified with the control sample. The PCR conditions were standardized and the amplified products were run on 2% agarose gel.
2.6. Validation and Gene Expression Study of Representative Genes FUB-1 and FUB-11
The mRNA expression levels were normalized to the level of housekeeping gene (18S rRNA) expression. The Ct values of the test samples were calculated and the data were expressed in terms of fold change over control sample.
2.7. Primer Efficiency and Calculations
The analysis for primer efficiency calculation is based on the following parameters: Five different dilutions of the sample (control) were taken for the analysis (1, 1:10, 1:100, 1:1000, and 1:10000). Mean CT value for the different dilutions was plotted in Y-axis against the log10 (1/Sample dilution) value in X-axis. The slope value was noted and used in the following equation for the efficiency % calculation: (10^(−1/The Slope Value).
3. RESULTS AND DISCUSSION
The present research work was concentrated on dual RNA-Seq analysis to compare the global gene expression patterns of pathogen infecting A. hypogaea. Alignment percentage of control, infected, and treated samples to A. hypogaea L. (93.35, 93.20, and 82.10%) was recorded, respectively [Table 7].
Reads of infected and treated sample that did not align to A. hypogaea were aligned with F. oxysporum genome and the number of reads recorded was 2,089,663 and 1,050,956, respectively [Table 8].
Next performed Blastx for our data with uniport database and the resulting top 5 abundant Gene ontology categories and Kyoto Encyclopaedia of Genes and Genomes (KEGG) pathways for significantly differential genes were shown in the form of Pie donut using High charts tool.
Pie-donut representation for Top 5 GO and Pathways w.r.t Pathogen for Treated versus infected for differential genes is depicted [Figure 1]. Heat Maps were created for Top 30 significantly up regulated and Top 30 significantly down regulated genes for treated versus infected. Tools used are Cluster 3.0 and Java TreeView (v 1.1.6). With Euclidean similarity matrix and average linkage rule [Figure 2].
HeatMaps were created using Cluster 3.0 and Java TreeView (v 1.1.6) for Top 30 significantly up regulated and Top 30 significantly downregulated genes for treated versus control [Figure 3].
After mapping, differential expression analysis was performed as “Treated vs. Infected,” after mapping, differentially expressed genes (DEG) (Pathogen Specific Expression) were recorded as 111 infected specific genes, 1162 treated, 33 up regulatory and 33 downregulatory [Table 1]. In this study, the analysis of region flanking the FUB gene cluster shows that the FUB cluster consisting of 12 genes of F. oxysporum infecting A. hypogaea L. Among them, FUB 1 and FUB 11 are expressed and are upregulated in A. hypogaea. Target gene/gene of interest was the FUB 1 and FUB 11. The genes under the study were first standardized and amplified with the control sample. The PCR conditions were standardized and the amplified products were further run on 2% agarose gel [Figure 4]. Gene expression study by RT PCR (CFX96 – Bio-Rad) was performed. The mRNA expression levels were normalized to the level of housekeeping gene (18S rRNA) expression. The Ct values of the test samples were calculated and the data were expressed in terms of fold change over control sample [Tables 2 and 3]. Primer efficiency for FUB-1, FUB-11, and housekeeping gene is shown [Tables 4-6]. The results show that the infected samples have higher expression of FUB-1 and FUB-11 than that of the treated samples when compared with the control sample [Figure 5].
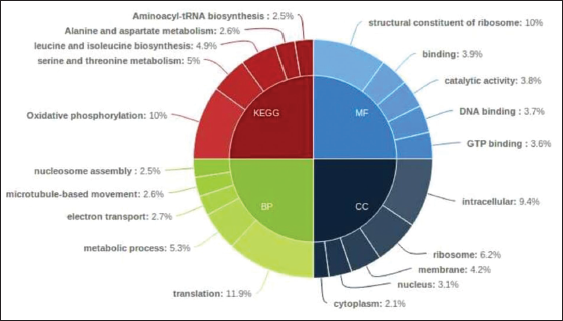 | Figure 1: Pie-donut representation for Top 5 GO and pathways w.r.t Pathogen for Treated versus infected for differential genes is depicted. [Click here to view] |
 | Figure 2: HeatMaps were created using Cluster 3.0 and Java TreeView (v 1.1.6) for Top 30 significantly upregulated and Top 30 significantly downregulated genes for treated versus infected. [Click here to view] |
 | Figure 3: HeatMaps were created using Cluster 3.0 and Java TreeView (v 1.1.6) for Top 30 significantly upregulated and Top 30 significantly downregulated genes for treated versus control. [Click here to view] |
 | Table 1: Differential expression analysis (pathogen?specific expression). [Click here to view] |
 | Table 2: Normalized FUB1 gene expression analysis of the test samples over control samples. [Click here to view] |
 | Table 3: Normalized FUB11 gene expression analysis of the test samples over control samples. [Click here to view] |
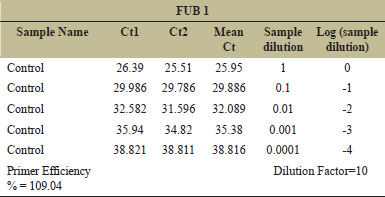 | Table 4: Primer efficiency for geneFUB?1. [Click here to view] |
 | Table 5: Primer efficiency for geneFUB?11. [Click here to view] |
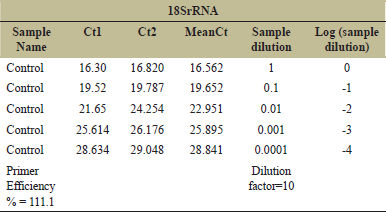 | Table 6: Primer efficiency for housekeeping gene (18SrRNA). [Click here to view] |
 | Figure 4: Polymerase chain reaction amplicons loaded on 2% agarose gel. [Click here to view] |
3.1. Lane Description
M: 100 bp ladder
1. FUB1- ~107bp
2. FUB11-~133bp
3. 18S rRNA.
CT value is generated by the software automatically.
Mean CT value is determined by: (CT1+CT2)/2
?CT value= Mean CT value of Housekeeping gene – Mean CT value of Target gene.
??CT value = ?CT value of control – ?CT value other samples.
This value is zero for the control sample and the difference for the infected and treated samples is provided.
Fold Change: 2^??CT value of the samples. This gives a value of 1 for the control samples whereas the samples show the fold changes value. This value is plotted in bar graph with target gene on the X-axis and fold change in Y-axis.
Bars represent standard errors from two independent biological experiments with three technical replicates each.
The present study concentrates on the RNA-Seq method for the identification of FUB genes responsible for FA production secreted by the F. oxysporum infecting A. hypogaea. The RNA-Seq method is an advanced technology used for gene expression studies as well as for the detection of pathogenicity and virulence factors. Gene ontology and pathways were found by Blastx which was hitting the infected and treated sequences against Uniprot_Fungus sequences, and top five GO and pathways w.r.t pathogen for treated versus infected differential genes that are depicted [Table 1].
According to the Blast (basic local alignment search tool) categories, the GO terms of F. oxysporum unigenes could be categorized into the following categories: Biological process, molecular function, and cellular component. As shown in Figure 1, unigenes can be allotted to one or more pathways. In the biological process category, unigenes were allotted to metabolic process (5.3%), cellular process (7.8%), and regulation of biological process (10%). In the cellular component category, unigenes were assigned to cell part (11.5%) and membrane part (4.2%) and both of them were highly represented. For the molecular function category, unigenes related to catalytic activity (3.8%), binding DNA (3.7%), and GTP binding (3.6%) were found to be the most abundant.
A high percentage of genes were assigned to the following categories: Post-translational modification, protein category, protein turnover, chaperones (3206, 13.17%), translation, ribosomal structure and biogenesis (2604, 10.69%), energy production and conversion (2014, 8.27%), and signal transduction mechanisms (1924, 7.90%).
Meanwhile, the KEGG database was used to search for active biochemical pathways in all unigenes of F. oxysporum. Carbohydrate metabolism mainly in 1,2-alpha-mannosidase activity (10%), amino acid metabolism (11.5%), energy metabolism (1651 unigenes), and lipid metabolism mainly in Benzoate degradation through hydroxylation; bile acid biosynthesis; fatty acid biosynthesis (path 2); and fatty acid metabolism were found. The above functional annotations showed that the clustered unigenes represented an extensive catalog encompassing large clustered unigenes expressed in F. oxysporum.
HeatMaps were created using Cluster 3.0 and Java Treeview (v 1.1.6) for top 30 significantly upregulated and top 30 significantly downregulated genes for treated versus infected [Figure 2] and also for treated versus control [Figure 3]. After mapping, DEG (pathogen specific expression) were recorded as 111 infected specific genes, 1162 treated, 33 up regulatory, and 33 down genes [Table 1]. In the present study, the analysis of region flanking the FUB gene cluster shows that the FUB cluster consisting of 12 genes of F. oxysporum infecting A. hypogaea L. The same was reported by [16] the person who reported that a FUB gene cluster consisting of 12 genes (FUB1–FUB12) was identified in different Fusarium species. The FUB cluster was identified with 12 genes and FUB-1 and FUB-11 are expressed and are upregulated in A. hypogaea L. FUB-1 is mainly involved in oxidoreductase activity (GO:0016491), catalytic activity (GO:0003824), and binding (GO:0005488), whereas FUB-11 is involved in transporter activity (GO:0005215). These genes were expressed and upregulated in relation to pathogenicity of F. oxysporum toward A. hypogaea L. Our results substantiated the earlier studies that the expression of FUB-1 in F. oxysporum was markedly upregulated during the early stages of plant infection and therefore tested the role of FA production in virulence [28]. The study describes upregulation of three gene from FUB gene cluster, namely, FUB11 that is presumed to encode an MFS transporter, FUB10 and FUB12 that are predicted to encode Zn(II)2Cys6 TF. Along with a Zn(II)2Cys6 TF, FUB12 is also predicted to contain a fungal TF regulatory domain. Thus, in addition to biosynthetic enzymes, the larger FUB cluster is predicted to encode proteins that could be involved in transport of FA across the cell membrane (FUB-11) and regulation of FUB gene expression (FUB-10 and FUB-12).
To investigate the expression profiles of two genes responsible for virulence in FA production during the infection process, RT-PCR analysis was carried out. The samples’ controlled, treated, and infected were analyzed for FUB1 and FUB11 genes expression. Total RNA was extracted from the test samples and cDNA was synthesized (reverse transcription). qPCR amplification was carried out using gene specific primers. The results were analyzed with control sample. The genes under the study were first standardized and amplified with the control sample. The PCR conditions were standardized and the amplified products were run on 2% agarose gel [Figure 4].
The above two FUB-1 and FUB-11 were confirmed by RT-PCR. Results in the present study revealed that FUB-1 showed a 1.1 fold and FUB-11 showed a 1.3 fold increase in expression during infection [Tables 2 and 3]. FUB-1 gene is primarily known to control FA production [11]. These are substantiated by the earlier reports which confirmed that FA was the first fungal phytotoxin and has key role in virulence [9,29], and several studies have provided circumstantial evidence linking FA production to plant pathogenicity [30].
The fold changes in the expression of the genes in infected and treated samples were plotted in Bar graph and represented [Figure 5] The findings demonstrated that the samples treated with T. viride and P. fluorescens reduced the toxicity of FA (0.1-fold increase in FUB-1 and 0.3-fold increase in FUB-11 expression) [31] also reported that a virulent strain of Pseudomonas solanacearum can detoxify FA in Fusarium wilt of tomato plants. The same is reported by the earlier workers [32] that the combined use of antagonistic mixtures have potential crop development benefits and more effective in suppressing the fungi causing disease.
 | Figure 5: Relative expression analysis of target genes (FUB1 and FUB11) and treated genes. [Click here to view] |
 | Table 7: Alignment to Arachis hypogaea L. genome. [Click here to view] |
 | Table 8: Alignment to Fusarium oxysporum genome. [Click here to view] |
4. CONCLUSION
The findings illustrate the recognition of FUB genes associated with the production of FA in F. oxysporum infecting A. hypogaea L. FUB-1 and FUB-11 are significantly expressed and are upregulated which confirmed that FA is fungal toxin playing a functional role in virulence in A. hypogaea. These findings illustrated the importance and shortcomings of plant genomic (transcriptomic) analyses in understanding the FUB genes associated with FA production in fungal pathogen. Trichoderma viride and P. fluorescens detoxify the FA indicating the potential benefits of using combination treatments to reduce FA formation during disease development and myotoxicity level at storage conditions. These findings yielded new sights to the molecular pathogenicity of F. oxysporum, and extending the groundnut resistance in over-expression of genes related to FA synthesis in A. hypogaea.
5. AUTHORSHIP
I, Dr P. Rajeswari as the corresponding and single author accept the four conditions of ICMJE that recommends the authorship.
6. ACKNOWLEDGMENTS
I am thankful to the University Grants Commission (UGC), India for funding. My Sincere Thanks to Roli and Kirti (Bionivid Technology) for helping me in performing transcriptomic analysis.
7. CONFLICTS OF INTEREST
The author declares that there is no conflict of interest.
8. FUNDING SOURCE
I am thankful to UGC University Grant Commission (India) for funding. Under the category of Post-Doctoral Fellowships for Women (F.15-1/2015-17/PDFWM-2015-17-AND-34147(SA-II).
REFERENCES
1. Leslie JF, Summerell BA. The Fusarium Laboratory Manual. Oxford: Blackwell Publishing; 2006.
2. Dean R, Van Kan JA, Pretorius ZA, Hammond-Kosack KE, Di Pietro A, Spanu PD, et al. The Top 10 fungal pathogens in molecular plant pathology. Mol Plant Pathol 2012;13:414-30.
3. Sultan M, Schulz MH, Richard H, Magen A, Klingenhoff A, Scherf M, et al. A global view of gene activity and alternative splicing by deep sequencing of the human transcriptome. Science 2008;321:956-60.
4. Filichkin SA, Priest HD, Givan SA, Shen R, Bryant DW, Fox SE, et al. Genome-wide mapping of alternative splicing in Arabidopsis thaliana. Genome Res 2010;20:45-58.
5. Severin AJ, Woody JL, Bolon YT, Joseph B, Diers BW, Farmer AD, et al. RNA-Seq Atlas of Glycine max: A guide to the soybean transcriptome. BMC Plant Biol 2010;10:160.
6. Ma LJ, van der Does HC, Borkovich KA, Coleman JJ, Daboussi MJ, Di Pietro A, et al. Comparative genomics reveals mobile pathogenicity chromosomes in Fusarium. Nature 2010;464:367-73.
7. Bacon CW, Porter JK, Norred WP, Leslie JF. Production of fusaric acid by Fusarium species. Appl Environ Microbiol 1996;62:4039-43.
8. Yabuta T, Kamb K, Hayashi T Biochemical studies of the “bakanae” fungus of rice. I. Fusarinic acid, a new product of the “Bakanae” fungus. J Agric Chem Soc Jpn 1937;10:1059-68.
9. Gaumann E. Fusaric acid as a wilt toxin. Phytopathology 1957;47:342-57.
10. Stipanovic RD, Puckhaber LS, Liu J, Bell AA. Phytotoxicity of fusaric acid and analogues to cotton. Toxicon 2011;57:176-78.
11. López-Díaz C, Rahjoo V, Sulyok M, Ghionna V, Martín-Vicente A, Capilla J, et al. Fusaric acid contributes to virulence of Fusarium oxysporum on plant and mammalian hosts. Mol Plant Pathol 2018;19:440-53.
12. Rajeswari P. Assessment of combination of biocontrol strains on the fusaric acid and other toxins secreted from Fusarium oxysporum by HPLC-MS/MS method and differential expression profiling in Arachis hypogaea L. Toxicol Int 2019;26:89-97.
13. Brown DW, Butchko RA, Busman M, Proctor RH. Identification of gene clusters associated with fusaric acid, fusarin, and perithecial pigment production in Fusarium verticillioides. Fungal Genet Biol 2012;49:521-32.
14. Niehaus EM, von Bargen KW, Espino JJ, Pfannmuller A, Humpf HU, Tudzynski B. Characterization of the fusaric acid gene cluster in Fusarium fujikuroi. Appl Microbiol Biotechnol 2014;98:1749-62.
15. Hoogendoorn K, Barra L, Waalwijk C, Dickschat JS, van der Lee TA, Medema MH. Evolution and diversity of biosynthetic gene clusters in Fusarium. Front Microbiol 2018;9:1158.
16. Studt L, Janevska S, Niehaus EM, Burkhardt I, Arndt B, Sieber CM, et al. Two separate key enzymes and two pathway-speci c transcription factors are involved in fusaric acid biosynthesis in Fusarium fujikuroi. Environ Microbiol 2016;18:936-56.
17. Keller NP, Turner G, Bennett JW. Fungal secondary metabolism-From biochemistry to genomics. Nat Rev Microbiol 2005;3:937-47.
18. Osbourn A. Secondary metabolic gene clusters: Evolutionary toolkits for chemical innovation. Trends Genet 2010;26:449-57.
19. Fullwood MJ, Wei CL, Liu ET, Ruan Y. Next-generation DNA sequencing of paired-end tags (PET) for transcriptome and genome analyses. Genome Res 2009;19:521-32.
20. Zenoni S, Ferrarini A, Giacomelli E, Xumerle L, Fasoli M, Malerba G, et al. Characterization of transcriptional complexity during berry development in Vitis vinifera Using RNA-Seq. Plant Physiol 2010;152:1787-9.
21. Peng Z, Liu F, Wang L, Zhou H, Paudel D, Tan L, et al. Transcriptome profiles reveal gene regulation of peanut (Arachis hypogaea L.) nodulation. Sci Rep 2017;7:40066.
22. Gamliel A, Katan T, Yunis H, Katan J. Fusarium wilt and crown rot of sweet basil: Involvement of soilborne and airborne inoculum. Phytopathology 1996;86:56-62.
23. Haas D, Blumer C, Keel C. Biocontrol ability of fluorescent pseudomonads genetically dissected: Importance of positive feed-back regulation. Biotechnology 2000;11:290-7.
24. Notz R, Maurhofer M, Dubach H, Haas D, Defago G. Fusric acid-producing strains of Fusarium oxysporum alter 2,4-diac- etylphloroglucinol biosynthetic gene expression in Pseudomonas Xuorescens CHA0 in vitro and in the rhizosphere of wheat. Appl Environ Microbiol 2002;68:2229-35.
25. Manjula K, Kishore GK, Girish AG, Singh SD. Combined application of Pseudomonas fluorescens and Trichoderma viride has an improved biocontrol activity against stem rot in groundnut. Plant Pathol J 2004;20:75-80.
26. Rajeswari P, Kapoor R. Combinatorial efficacy of Trichoderma spp. and Pseudomonas fluorescens to enhance suppression of cell wall degrading enzymes produced by Fusarium wilt of Arachis hypogaea L. Int J Agric Res Innov Technol 2017;7:36-42.
27. Trapnell C, Pachter L, Salzberg SL. TopHat: Discovering splice junctions with RNA-Seq. Bioinformatics 2009;25:1105-11.
28. López-Díaz VC, Rahjoo V, Sulyok M, Ghionna V, Marttin-Vicente A, Matin-Vicente A, et al. Fusaric acid contributes to virulence of Fusarium oxysporum on plant and mammalian hosts. Mol Plant Pathol 2018;19:440-53.
29. Gaumann E. The mechanism of fusaric acid injury. Phytopathology 1958;48:670-86.
30. Dong X, Xiong Y, Ling N, Shen Q, Guo S. Fusaric acid accelerates the senescence of leaf in banana when infected by Fusarium. World J Microbiol Biotechnol 2014;30:1399-408.
31. Toyoda H, Hashimoto H, Utsumi R, Kobayashi H, Ouchi S. Detoxification of fusaric acid by a fusaric acid-resistant mutant of Pseudomonas solanacearum and its application to biological control Fusarium wilt of tomato. Phytopathology 1988;78:1307-11.
32. Eshel D, Regev R, Orenstein J, Droby S, Gan-Mor S. Combining physical, chemical and biological methods for synergistic control of postharvest diseases: A case study of black root rot of carrot. Postharvest Biol Technol 2009;54:48-52.