
1. INTRODUCTION
Xylan is a polysaccharide found abundantly in hemicelluloses and is involved in cross-linking cellulose fibers in the cell wall of plants. To ensure the ease of the process, xylan content needs to be reduced, especially in industries such as animal feed (removal of xylan to improve the nutritive value of feed), paper pulp (for ease of bleaching lignin), and plant oils (to increase the availability of bound fat) [1].
Xylanases are enzymes that degrade β-1, 4-glycosidic linkage of the xylan fibers. These enzymes along with xylosidases convert the xylan fibers to xylose subunits [1–5]. Among the xylanase degrading enzymes, only a few glycoside families are monofunctional with different pH, working temperatures and these find greater applications in industries, with xylanases in acidic pH used in animal feed and baking industry [6], neutral to alkaline xylanases in paper recycling and paper pulp processing industries [1,3,7]. Among xylanases, GH11 and GH10 xylanases find particular applications in paper pulp industries, where the removal of xylan aids in delignification of paper pulp and reduces chlorine consumption in the bleach step, reducing the production of organic halides [1,3,7].
To produce the enzyme in bulk at economically viable production costs, we engineered the xylanase gene expressing GH11 family xylanase to express in a host organism such as Pichia pastoris which is more suited for submerged fermentation conditions. Pichia pastoris is methylotrophic yeast which is a successful system for the production of a wide variety of recombinant proteins [3,7,8]. This expression system offers greater advantages such as well studied genome [9,10], tight promoter [alcohol oxidase promoter1 (AOX1)], grows to greater cell density [10], has been used to successfully express economically important proteins such as phytase, catalase L, tumor necrosis factor, tetanus toxin fragment C, α-amylase, to name a few [11].
In this study, the Pichia expression system was used as a host to express GH11 xylanase from Bacillus pumilus SSP-34 and the enzyme was purified and characterized.
2. MATERIALS AND METHODS
2.1. Strains, Plasmids and Media
Bacillus pumilus SSP-34 [Microbial Type Culture collection and gene bank (MTCC) 5015] was purchased from CSIR-National Institute for Interdisciplinary Science and Technology (NIIST) (formerly RRL, Trivandrum). Bacillus pumilus was grown on xylan media (0.5% oat spelt xylan, 0.1% KH2PO4, 0.02% MgSO4, 0.5% yeast extract, 0.5% peptone and 2.5% agar) at 30°C for 24 hours. Escherichia coli TOP10F (Invitrogen) was used to maintain and construct plasmids used in this study. Escherichia coli TOP10F was grown on Luria Bertani broth (LB) media (1% tryptone, 0.5% yeast extract, 1% NaCl with pH 7.0) at 37°C for 24 hours. Pichia pastoris GS115 (Invitrogen) was grown and maintained on Yeast extract Peptone Dextrose (YPD) media (1% yeast extract, 2% peptone, and 2% dextrose).
The expression vector used to express the constructed gene in P. pastoris was pPIC3.5K (Invitrogen) and the α-factor mating signal sequence was derived from the pPIC9K sequence (Invitrogen).
2.2. Assay of Xylanase
Xylanase assay was carried out as described by Poorna [12]. 0.1 ml of the suitably diluted enzyme (containing 1–5 μg of protein) was added to the pre-heated substrate (0.5% beechwood xylan prepared in 0.1 M phosphate buffer, pH 7.0). The enzyme assay was carried out at 50°C for 10 minutes. The reaction mixture was heated in a water bath for 5 minutes after the addition of 1.5 ml of dinitrosalicylic acid (DNS) solution. The absorbance of the mixture was measured at 540 nm using Eppendorf biospectrophotometer. The amount of reducing sugar released was calculated using a xylose standard. One unit of xylanase activity is defined as the amount of enzyme required to liberate 1 μmol of reducing sugar equivalent to xylose, per minute, under standard assay conditions. Total soluble proteins were estimated by Lowry’s et al. [13] method, using bovine serum albumin as the standard.
2.3. Gene Design and Plasmid Construction
To optimize the gene for expression in P. pastoris, the nucleotide sequence (NCBI accession number: AY887130.1) of the protein (Uniprot ID: Q5EFR9) was optimized using the Integrated DNA technologies (IDT) dna codon optimization tool [14]. The native signal peptide from B. pumilus was replaced with α-factor mating signal sequence from P. pastoris GS115.
The gene was synthesized by IDTdna, Belgium. The synthesized gene (α-xynA, NCBI accession number:MW691224) was amplified using xynCFP (5?-AGATGGATCCAAACGATG-3?) and xynCRP (5?-CTCATCACTAATTGCCTATG-3?) primers. Polymerase Chain Reaction (PCR) mastermix was prepared using 0.5 μM of primers dissolved in double distilled water (ddH2O), 1.8 M betaine, 1× High fidelity buffer (HF buffer), 0.25 μl Phusion polymerase [New England Biolabs (NEB)], and 0.5 μl of Deoxy Nucleotide TriPhosphate (dNTPs) per 25 μl of PCR master mix. The PCR parameters were as follows: denaturation at 98°C for 30 seconds, annealing step at 49°C for 10 seconds, extension at 72°C for 40 seconds, and the PCR parameters were cycled for 32 cycles. The bands were confirmed using gel electrophoresis using 1.2% agarose in Tris-acetate-ethylene diamine tetra acetic acid (EDTA) buffer.
Overhangs to the gene were added using primers xyngFP (5?-aaacaactaattattcgaagAGATGGATCCAAACGATGC-3?) and xyngRP (5?-tctaaggcgaattaattcgcCTCATCACTAATTGCCTATGAATAATTG-3?) using the same PCR conditions mentioned above. 2 μl of purified PCR product (gene with overhangs) was mixed with 4 μl of pPIC3.5K (linearized using BamHI and SnaBI restriction endonucleases) along with Gibson assembly mastermix (NEB) and incubated at 50°C for 30 minutes. The reaction mixture was diluted with ddH2O to 1:10 ratio and 1 μl of the diluted mixture was used to transform competent E. coli TOP10F cells. The transformed cells were confirmed by colony PCR using 5?AOX1 (5?-GACTGGTTCCAATTGACAAGC-3?) and 3?AOX1 (5?-GCAAATGGCATTCTGACATCC-3?) primers.
2.4. Transformation into P. pastoris GS115
The competent cells were electroporated with pPIC3.5K/α-xynA (concentration 1.4 μg/μl), linearized using SacI (Thermofisher Scientific). The transformants were grown on YPD geneticin (G418) plates at 30°C for 3 days. Gene copy number was roughly estimated by growing the transformants on YPD media with varying concentrations (1X–9X) of geneticin (where X is 0.25 mg/ml). Transformants from YPD geneticin plates (4X–9X) were subcultured on Buffered-methanol-complex media (BMMY) agar plates (1% yeast extract, 1% peptone, 100 mM phosphate buffer pH 6.0, 1.34% yeast nitrogenous base, 4 × 10−5% biotin and 1% methanol) containing 1% oat spelt xylan. Clear zones in the BMMY plates were used to select the colonies expressing the xylanase enzyme. These colonies expressing greater enzyme yields were confirmed by xylanase assay (DNS method).
2.5. Fermentation and Downstream Processing (B. pumilus)
Xylanase from B. pumilus was produced using solid-state fermentation (xylan media, containing wheat bran with 2% Na2CO3, 0.1% K2HPO4 and 0.04% MgSO4). Wheat bran media was added with water in the ratio of 1:2 (w/v). The media was inoculated with fresh inoculum and incubated at 30°C for 120 hours. The secreted enzyme was extracted in 8% NaCl solution. The extract was centrifuged at 7,000 g to separate debris and the supernatant was salted out using 100% ammonium sulfate saturation, with constant stirring at 4°C for 6 hours. The precipitate from salting-out was collected by centrifugation and dispersed in 50 mM phosphate buffer, pH 7.0.
2.6. Fermentation and Downstream Processing (P. pastoris)
To produce xylanase enzyme, P. pastoris was grown under submerged fermentation conditions, on a minimal media (0.0093% CaSO4, 0.182% K2SO4, 0.149% MgSO4, 0.0413% KOH, 0.267% H3PO4, 2% glycerol, pH 5.5). Media pH was set and maintained at pH 5.5 during the growth phase and pH 6.0 during the enzyme production phase. 5% (v/v) Inoculum (16 hours flask culture) was added to the media and fermentation was carried out at 30°C for around 16 hours or until the optical density at 600 nm (OD600) reaches between 60 and 70. During the entire fermentation process, dissolved oxygen level (DO) was carefully monitored to ensure the healthy growth of the culture. An increase in DO values was used as an indicator for starvation due to insufficient nutrients. Once the desired OD (OD600 of 100) was achieved, the culture was starved-off glycerol for 1–2 hours, after which methanol was fed for protein expression for 96 hours or until the media reached an OD600 of 200. Methanol was fed at the rate of 2.5 ml/hour/l for an initial 24 hours, followed by 6.0 ml/hour/l for the next 24 hours and subsequently 8 ml/hour/l for the next 24 hours and 12 ml/hour/l for the last phase.
After fermentation, the broth was centrifuged to remove the cell pellet. The supernatant was precipitated using 100% ammonium sulfate saturation with constant stirring at 4°C for 6 hours. The enzyme precipitate was collected by centrifugation and dispersed in 50 mM phosphate buffer, pH 7.0.
2.7. Purification of Xylanases
The ammonium sulfate pellet samples from both native and recombinant cultures were dialyzed against phosphate buffer and applied to diethyl amino ethyl (DEAE) cellulose ion exchange column, equilibrated with 50 mM phosphate buffer pH 7.0. Elution was carried out using 0 to 1 M NaCl. The active fractions were pooled, concentrated using viva-spin columns (GE healthcare). Using 4% agarose gels (running buffer was Tris-glycine buffer pH 8.3), horizontal electrophoresis was carried out. The xylanase band position was identified (by zymogram), excised and the enzyme was eluted into 50 mM phosphate buffer pH 7.0. The eluted enzyme was concentrated using a viva-spin column and used for further experiments.
2.8. Sodium Dodecyl Sulphate Polyacrylamide Gel Electrophoresis (SDS-PAGE)
SDS-PAGE was carried out with 10% polyacrylamide gel as described by Lammeli [15]. Silver staining was used to visualize the protein band pattern. The apparent molecular weight of the purified xylanase was estimated using gel reader software (Image Lab, Gel Doc EZ Imager, BioRad).
2.9. Zymogram
Horizontal agarose electrophoresis was carried out using a non-denatured sample with ribonuclease and lysozyme as standards. After the run, the gel was washed in 50 mM phosphate buffer pH 7.0, placed on 0.5% xylan-agar plate prepared in the same buffer and incubated at 40°C for 30 minutes. Congo red (0.1%) was used to stain the gel and the excess stain was removed by washing with 1 M NaCl. The clear zones were observed under a gel reader (BioRad).
2.10. Protein Sequencing by Liquid chromatography-mass spectrometry (LC-MS)/MS
The protein samples were initially resolved on SDS-PAGE and stained using Coomassie brilliant blue. The resolved bands were excised from the gel, de-stained using 100 mM ammonium bicarbonate/acetonitrile (1:1) solution, followed by acetonitrile wash. The samples were digested using trypsin and the tryptic digest of the proteins was resolved on the nano-High pressure liquid chromatography (HPLC) column (1200, 1D nano-LC, Agilent) using acetonitrile/formic acid gradient. The peptides were ionized by an automated Electron Spray Ionisation (ESI) ESI-chip (NanomateTriversa, Advion) and the generated ions were transferred to the mass spectrometer (LTQ-Orbitrap Discovery, Thermo Scientific) through an LC coupler. The peptide data obtained from the mass spectrometer were used to establish the identity of the proteins and the protein sequence was later reconstructed using MASCOT and PEAKS software.
2.11. Effect of pH and Temperature on Xylanase activity
Optimum pH for activity was determined by carrying out assays at different pH ranging from pH 4.0 to 11.0. Buffers used were 100 mM acetate buffer for pH 4.0–5.5, phosphate buffer for pH 6–8 and glycine-NaOH buffer for pH 8.5–11.0. The assay was carried out at 50°C. The optimum temperature for activity at pH 7.0 was evaluated by assaying at different temperatures (20°C–80°C). The highest activity obtained was taken as 100% and expressed as percent activity.
Thermostability was evaluated by incubating the enzyme at different temperatures (45°C and 55°C) for 60 minutes. Samples were drawn every 10 minutes and cooled on an ice bath for 5 minutes. The residual activity in the samples was determined by assaying at 50°C, pH 7.0. Control was taken as 100% activity and test samples were expressed as percent residual activity.
2.12. Kinetic Parameters of the Enzymes
The Km and Vmax values of xylanase were determined using beechwood xylan as substrate (concentration range 0%–1.5%). The assay was carried out for 10 minutes at pH 7.0 and 50°C. Km and Vmax values were derived from the Lineweaver–Burk plot.
3. RESULTS
3.1. Bioinformatic Analysis
The native gene sequence (NCBI accession number: AY887130.1) indicated that the gene was 687 bp long and consisted of a signal peptide and the protein-coding regions and a gap between the two regions. The signal peptide region was separated from the xylanase coding region through a stable dyad symmetry or a hairpin loop (stable at 78°C) using mfold software [16]. The signal peptide region had a Guanine-Cytosine content (GC content) of 28%, whereas the protein-coding region had a GC content of 63%. To express the protein in Pichia, the sequence was modified to replace signal peptide with an α-factor mating signal peptide, codon-optimized for better protein expression and to eliminate the effects of DNA secondary structure during Pichia transformation. The two sequences are given in Figure 1.
3.2. Codon Optimization
The first half of the native sequence was replaced with codons of higher usage, while the rest of the sequence was replaced with codons of lower usage as per the Pichia codon usage table. Care was taken to reduce the overall GC content to 42%. The sequence after attaching with alpha mating signal peptide was used to synthesize the gene for expression in P. pastoris GS115. The gene sequence was checked for dyad symmetry and special care was taken to remove secondary structures by replacing them with appropriate codons. The final gene size was 876 bp. Electroporation was the method of choice for transforming the codon-optimized gene to Pichia cells.
 | Figure 1: A) DNA alignment of native enzyme (Uniprot ID: Q5EFR9, NCBI accession number AY887130.1) to the codon-optimized sequence. B) Vector map of the constructed plasmid carrying the α-xynA gene. C) Agarose gel electrophoresis: GeneRuler 1 kb plus marker (lane M), colony PCR using sequencing primers 5?AOX1 forward primer and 3?AOX1 reverse primer (lane 1). [Click here to view] |
3.3. Screening Colonies
Transformants from YPD geneticin plates were further screened on BMMY agar plates for xylanase expression (seen as clear zones on the plate). The colonies were further selected based on the expression levels of the protein by DNS assay.
3.4. Fermentation Yields
The composition of the media and the fermentation of the native and recombinant protein are outlined in materials and methods. Fermentation of B. pumilus yielded xylanase activity of 10,000 U/g at the end of 120 hours, while submerged fermentation of P. pastoris yielded 6,000 U/ml at the end of 120 hours.
3.5. Purification of Xylanases
GH11 Xylanases from both the culture media were salted out using 100% ammonium sulfate saturation, desalted by dialysis and separated by DEAE-cellulose anion-exchange chromatography. Both the native and recombinant enzymes were eluted in the void volume with a specific activity of 826 and 1,137 U/mg, respectively (Fig. 2a and b). Further purification of the enzyme was carried out using agarose electrophoresis at pH 8.3. Xylanase enzyme, with pI: 9.05 (pI calculator, Expasy) is positively charged at the pH used (running buffer pH 8.3) and hence moved toward the cathode. The DEAE pool was concentrated using viva-spin column and horizontal electrophoresis was carried out using 4% agarose. The sample wells were placed at the center of the gel. Xylanase bands that moved towards the cathode were identified by zymogram using substrate gel (oat spelt xylan). The xylanase bands were then excised and the enzyme was eluted into the buffer, concentrated and identified on SDS-PAGE. The purified xylanases from B. pumilus and P. pastoris had a specific activity of 937 U/mg and 1,268 U/mg, respectively (Table 1a and b). The recombinant xylanase from P. pastoris had slightly higher specific activity when compared with the native xylanase.
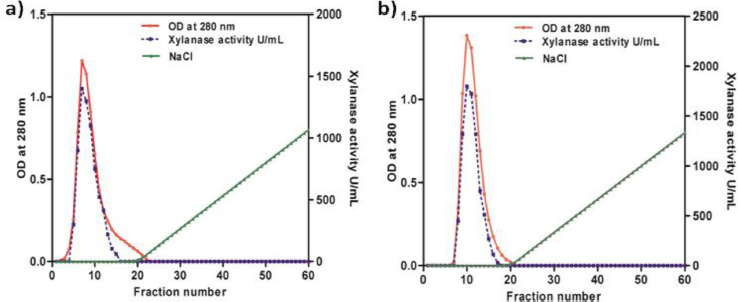 | Figure 2: Fast performance liquid chromatography data of (a) native GH11 xylanase from B. pumilus and (b) recombinant xylanase expressed in P. pastoris GS115. [Click here to view] |
 | Table 1: (a and b) Purification of xylanase from B. pumilus SSP-34. Purification of xylanase from P. pastoris GS115. [Click here to view] |
3.6. Sodium Dodecyl Sulphate Polyacrylamide Gel Electrophoresis
The purified native xylanase and recombinant enzyme moved asa single band in SDS-PAGE (Fig. 3a). The molecular weights of native and recombinant xylanase were determined to be 21.2 ± 0.5 and 24.8 ± 0.4 kDa, respectively. The apparent increase in the molecular weight of recombinant xylanase could be due to glycosylation of the protein molecule which is common in yeasts. The increase in the molecular weight was also evident by the horizontal agarose electrophoresis and zymogram (Fig. 3b and 3c).
3.7. Protein Sequence
The sequence of the native protein is given in Figure 1, with a molecular weight of 22.5 kDa and had 201 residues (without signal peptide). This sequence was in agreement with the earlier reported protein sequence of a GH11 xylanase from B. pumilus except for N-terminal residue (alanine being replaced by arginine) [17]. The protein sequence was also confirmed by LC-MS protein sequencing data (Fig. 4).
3.8. Effect of pH and Temperature on Activity
The pH optima and temperature optima for activity were measured for both native and recombinant protein (Fig. 5a and 5b). The native xylanase had a pH optimum of 7.0 and the recombinant enzyme had a pH optimum of 6.5. The native xylanase was most active between pH 5.5–9.0 and recombinant protein was most active between pH 5.0–8.5. There was a slight shift in the pH optimum of the recombinant enzyme. The temperature optima for both native and recombinant enzymes were at 50°C. The recombinant enzyme had a wider working range with substantially higher activity at 30°C and 40°C working temperature in comparison to the native enzyme.
 | Figure 3: Electrophoretic analysis of xylanase enzyme (a) Silver-stained PAGE gel of native and recombinant xylanases, (b) Horizontal electrophoresis gel of native and recombinant xylanases along with reference proteins and (c) Zymogram of native and recombinant xylanases was carried out using xylan-agar and staining was carried out using congo red stain. [Click here to view] |
 | Figure 4: Amino acid sequence of the native GH11 xylanase from B. pumilus. [Click here to view] |
3.9. Thermostability of Native and Recombinant Xylanases
The thermostability measurements showed that the recombinant enzyme was slightly more stable than the native enzyme with Tm of 55°C and 53°C, respectively. Both the enzymes were stable up to 50°C (Fig. 5b). The thermostability of the native and recombinant xylanases was measured at 45°C and 55°C. The rate constant k was 0.067 and 0.035 minute−1 and the half-life of the enzymes was 10.5 and 20.9 minutes at 55°C for native and recombinant xylanases respectively (Fig. 5c and 5d). The recombinant enzyme was more stable in comparison to the native enzyme.
3.10. Kinetic Parameters of the Enzymes
The Km and Vmax values for the native and recombinant enzymes were similar. Km was 20 mg/ml for both native and recombinant xylanases indicating the structural similarity of the two enzymes, Vmax values were 937 and 1,268 U/mg and Kcat values were 1,415 × 103 and 1,650 × 103 M/mg/minute for native and recombinant xylanases, respectively (Fig. 6).
4. DISCUSSION
GH11 xylanase finds greater applications in paper pulp bio-bleaching and paper recycling industries. As such, many of the GH11 alkaline-neutral pH enzymes can be used to reduce chlorine usage in the bleaching step of the process. The current GH11 xylanase was derived from B. pumilus, a soil-dwelling generally regarded as safe microorganism, using solid-state fermentation. Due to difficulties in handling the fermentation and successes reported in expressing heterologous proteins [3,7,8], P. pastoris, a methylotrophic yeast was used as the expression system. The ease of handling cultures, its ability to grow to a greater cell density, a tightly regulated promoter system (AOX1 promoter) and the ability to use methanol (toxic for other microorganisms and cheaper inducer) makes Pichia an attractive host for expressing GH11 xylanase as achieved in this current study.
In this study, we designed a codon-optimized gene, encoding the GH11 xylanase containing α-factor mating signal sequence for greater expression and secretion in the Pichia host (Fig. 1). Yield comparisons of native and recombinant proteins indicated greater expression levels of the recombinant protein. This makes it conducible for bulk production of the enzyme for industrial applications.
 | Figure 5: Comparison of native and recombinant GH11 xylanases a) pH profileplot b) Activity versus temperature plot. c) Half-life analysis of native and recombinant xylanases at temperatures 45°C and 55°C. d) Log % residual activity versus time plot of native and recombinant xylanases at 55°C. [Click here to view] |
 | Figure 6: Lineweaver-Burk plot comparison of native and recombinant GH11 xylanases. [Click here to view] |
We were able to transform the codon-optimized gene by the electroporation method. The other methods that have been used for the transformation of the xynA gene were PEG1000 induced transformation [18] and the electroporation method [19].
The molecular weights of the recombinant protein and native protein were 25 and 22 kDa, respectively. The apparent increase in molecular weight could be due to glycosylation [20]. A similar increase in molecular weight of the expressed protein in P. pastoris has been reported [21]. Based on the amino acid sequence alignment, there are four probable glycosylation sites on GH11 xylanase (NetNGlyc 1.0 Server) [22].
The expressed protein was purified to homogeneity by ammonium sulfate precipitation followed by anion exchange chromatography and subsequently purified to homogeneity. The amino acid sequence of the native enzyme was given in the Figure 1. This is a basic protein with a ratio of acidic to basic amino acid (13:20), and the theoretical pI was 9.05. The recombinant protein had higher specific activity compared to the native. The kinetic parameters like Km and Vmax, pHopt and Topt were comparable. The reported Km and Vmax values for similar systems are 6.5 mg/ml on Oat spelt xylan [17], 4 mg/ml on Oat spelt xylan [12,23]. The temperature stability and the molecular weight of the recombinant enzyme were slightly different. This difference in temperature stability and molecular weight could be due to the glycosylation of the recombinant protein. Similar observations have been made by Teng et al. [20]. The enhanced stability of the recombinant protein makes it a preferred choice for industrial applications.
The expression of the GH11 xylanase from B. pumilus in P. pastoris opens the possibilities of creating mutants with improved thermal stability, improved activity, and designer pH stability. It may be pertinent to mention our in-silico studies for the improved stability of GH11 xylanase [24]. One of the notable features of the GH11 xylanase from B. pumilus is the contribution of hydrophobic sidechains to the stability of the protein.
5. CONCLUSION
The codon-optimized xynA gene coding for GH11 xylanase from B. pumilus was modified to replace the signaling factor with Pichia expression system. 876 bp gene was synthesized and expressed in methylotrophic yeast P. pastoris GS115. The recombinant protein was purified, characterized for enzymatic properties and thermal stability. Recombinant xylanase was more thermostable and had higher molecular weight probably due to glycosylation.
6. ACKNOWLEDGMENT
The authors gratefully acknowledge the financial support and microbial cultures from KAYPEEYES Biotech Private limited, Mysuru, India. The authors thank Mr. Krishna Bhat Kadappu, Managing Director and staff of KAYPEEYES Biotech private limited, Mysuru, India, for their guidance while carrying out the research. The inputs provided by C-Camp (Hebbal, Bengaluru, India) towards protein sequencing is gratefully acknowledged. The authors are extremely thankful to Dr. Rajeev Kumar Sukumaran, Principal Scientist and Head, Microbial Processes and Technology, CSIR-NIIST, Trivandrum, India, for providing the B. pumilus SSP-34 identification number.
7. LIST OF ABBREVIATIONS
AOX1 Alcohol oxidase promoter1
BMMY Buffered-methanol-complex media
DEAE celluloseDiethyl amino ethyl cellulose
DNS Dinitro salicylic acid
DO Dissolved oxygen
GH11/GH12 Glycoside hydrolase 11/12
SDS-PAGE Sodium dodecyl sulphate polyacrylamide gel electrophoresis
8. CONFLICTS OF INTEREST
The authors declare no conflict of interest in this work.
9. FUNDING
There is no funding to report.
10. ETHICS APPROVAL
This article does not contain any studies with human participants or animals performed by any of the authors.
11. AUTHOR CONTRIBUTIONS
KRK and AGA conceived and supervised the project, designed the experiments, critically evaluated, edited, and approved the final manuscript, SKB and KP performed the experiments, analyzed the data, and drafted the manuscript.
REFERENCES
1. Collins T, Gerday C, Feller G. Xylanases, xylanase families and extremophilic xylanases. FEMS Microbiol Rev 2005;29(1):3–23; doi:10.1016/j.femsre.2004.06.005
2. Biely P, Puls J, Schneider H. Acetyl xylan esterases in fungal cellulolytic systems. FEBS Lett 1985;186(1):80–4; doi:10.1016/0014-5793(85)81343-0
3. Cheng YF, Yang CH, Liu, WH. Cloning and expression of Thermobifida xylanase gene in the methylotrophic yeast Pichia pastoris. Enzyme Microb Technol 2005;37(5):541–6; doi:10.1016/j.enzmictec.2005.04.006
4. Paës G, Berrin JG, Beaugrand J. GH11 xylanases: structure/function/properties relationships and applications. Biotechnol Ad 2012;30(3):564–92; doi:10.1016/j.biotechadv.2011.10.003
5. Wang K, Luo H, Tia J, Turunen O, Huang H, Shi, et al. Thermostability improvement of a Streptomyces xylanase by introducing proline and glutamic acid residues. Appl Environ Microbiol 2014;80(7):2158–65; doi:10.1128/AEM.03458-13
6. Singh AB. Production, characteristics, and biotechnological applications of microbial xylanases. Appl Microbiol Biotechnol 2019;103(21–22):8763–84; doi:10.1007/s00253-019-10108-6
7. Damas CT, Almeida MS, Kurtenbach E, Martins OB, Pereira N, Andrade CMMC, et al. Optimized expression of a thermostable xylanase from Thermomyces lanuginosus in Pichia pastoris. Appl Environ Microbiol 2003;69(10):6064–72; doi:10.1128/AEM.69.10.6064
8. Cregg JM, Cereghino JL, Shi J, Higgins DR. Recombinant protein expression in Pichia pastoris. Mol Biotechnol 2000;16(1):23–52; doi:10.1385/MB:16:1:23
9. He J, Yu B, Zhang K, Ding X, ChenD. Expression of endo-1, 4-beta-xylanase from Trichoderma reesei in Pichia pastoris and functional characterization of the produced enzyme. BMC Biotechnol 2009;9(56):1–10; doi:10.1186/1472-6750-9-56
10. Jin X, Meng N, Xia LM. Expression of an endo-β-1,4-glucanase gene from Orpinomyces PC-2 in Pichia pastoris. Int J Mol Sci 2011;12(5):3366–80; doi:10.3390/ijms12053366
11. Clare JJ, Rayment FB, Ballantine SP, Sreekrishna K, Romanos MA. High-level expression of tetanus toxin fragment C in Pichia pastoris strains containing multiple tandem integrations of the gene. Biotechnol 1991;9(5):455–60; doi:10.1038/nbt0591-455
12. Poorna CA. Purification and biochemical characterization of xylanases from Bacillus pumilus and their potential for hydrolysis of polysaccharides. Ferment Techol 2011;1(1):1–5; doi:10.4172/2167-7972
13. Lowry OH, Rosenbrough NJ, Farr AL, Randall RJ. Protein measurement with the folin phenol reagent. J Biol Chem 1951;193(1):265–75. CrossRef
14. Sebesta J, Peebles CA. Improving heterologous protein expression in Synechocystis sp. PCC 6803 for alpha-bisabolene production. Metab Eng Commun 2020;10(e0017):1–9; doi:10.1016/j.mec.2019.e00117
15. Lammeli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 1970;277(5259):680–5; doi:10.1038/227680a0
16. Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res 2003;31(13):3406–15; doi:10.1093/nar/gkg595
17. Subramaniyan S, Prema P. Cellulase-free xylanases from Bacillus and other microorganisms. FEMS Microbiol Lett 2000;183(1):1–7; doi:10.1016/S0378-1097(99)00620-5
18. Zhang GM, Hu Y, Zhuang YH, Ma LX, Zhang XE. Molecular cloning and heterologous expression of an alkaline xylanase from Bacillus pumilus HBP8 in Pichia pastoris. Biocatal Biotransform 2006;24(5):371–9; doi:10.1080/10242420600768771
19. Zheng W. Enhancement of heterogeneous alkaline xylanase production in Pichia pastoris GS115. AIP Conf Proc 2017;1864(1):1–4; doi:10.1063/1.4992906
20. Teng D, Fan Y, Yang YL, Tian ZG, Luo J, Wang JH. Codon optimization of Bacillus licheniformis β-1,3-1,4-glucanase gene and its expression in Pichia pastoris. Appl Microbiol Biotechnol 2007;74(5):1074–83; doi:10.1007/s00253-006-0765-z
21. Bretthauer RK, Castellino FJ. Glycosylation of Pichia pastoris-derived proteins. Biotechnol Appl Biochem 1999;30(3):193–200; doi:10.1111/j.1470-8744.1999.tb00770.x
22. Gupta, R, Brunak S. Prediction of glycosylation across the human proteome and the correlation to protein function. Pacific Symp Biocomput 2002:310–322; doi:10.1142/9789812799623_0029
23. Nakamura M, Nagamine T, Takenaka A, Aminov RI, Ogata K. Molecular cloning, nucleotide sequence and characteristics of a xylanase gene (xyn A) from Ruminococcus albus 7. Anim Sci J 2002;73(5):347–52; doi:10.1046/j.1344-3941.2002.00048.x
24. Bhat SK, Purushothaman K, Kini KR, Rao AG. Design of mutants of GH11 xylanase from Bacillus pumilus for enhanced stability by amino acid substitutions in the N-terminal region: an in silico analysis. J Biomol Struct Dyn 2021:1–14; doi:10.1080/07391102.2021.1899988