
1. INTRODUCTION
The Mycobacterium tuberculosis (MTB) bacterium causes TB. Recently in 2020, 1.3 million people died from Tuberculosis (TB) disease. The effects of TB are observed in different age groups worldwide, mostly in young, adults, and people living in developing countries [1]. In past years, various antibiotics drugs utilized to treat, and fight against TB, but some strains have become resistant to drugs [2]. TB is challenging to diagnose using clinical, radiologic, bacteriological, and histological methods [3]. At present, the treatment of TB patients relies on clinical and laboratory evaluations, which have a number of limitations; the standardized methods do not account the individual variability in pathogenesis of MTB [4]. TB is treatable and curable but the fatality rate can be reduced only if we identify the potential drug targets where a bacterium is failed to develop resistance to the novel drug. The future treatment of TB patients may be improved using many systems biology domains. Next-generation sequencing and whole-genome sequencing methods provides a diagnostic role for treating patients with the disease [5,6]. Pharmacokinetic and pharmacodynamic techniques combined with systems biology have the potential to highlight the issues. Our understanding of the route of action of antibiotics is incorporated into systems pharmacology-based pharmacodynamic models to predict antibiotic effects on bacteria, for example, drug-target interactions [7]. High-throughput gene expression data made it possible to create large-scale gene regulatory networks. Gene regulatory networks provide as closer look to the clinical and medical application and can be seen as a bottleneck between the genotype and the phenotypes. Hence, it is important to understand the communication that occurs within MTB itself and with the host through molecular interaction. Two-component systems (TCSs) play an important role in bacteria which is ubiquitous and also important for cell signaling processes such as cell communications and cell adaptations [8]. The absence of TCS proteins in humans and other mammals’ cellular systems makes them important drug targets potential. MTB has 11 pairs of TCS to perform its regular function and they are potentially attractive drug targets [9]. Understanding the pathways affected by these systems would unfurl effective measures for preventing MTB multiplications that ultimately result in pathogenesis. The MtrAB system is one of the two essential TCS in MTB, wherein MtrB is the sensor kinase, and MtrA is the cognate response regulator [10]. The MtrAB system is associated with replication, cell division, and cell wall formation. The MtrAB system is available in all mycobacterial species but is essential in MTB only for viability [10,11]. The virulence and infection mechanisms of MTB require signal transduction and one of the key biological processes is protein-protein interaction (PPI), which is a tool to analyze signal transduction.
The system biology approach using PPIs provided the key to understanding complex biological systems and helps to understand the importance of key regulators [12]. The network theory-based analysis helps to understand the topological properties of complex systems [13]. The modules in the hierarchical network are of particular interest because modules many correspond to independent functions [14,15]. The hub protein controls the stability of the network as well as modules [16-18]. Hence, the interaction between the hub and modules is crucial for network stability and communication. It was pertinent, therefore, to understand the communication within MTB and with its host based on MtrAB using a network biology approach.
2. METHODS
2.1. Construction and Statistical Analysis of MtrAB PPI Network
The PPIN of MtrA and MtrB genes was constructed using the STRING (Szklarczyk et al., 2019) database. The PPI information of MtrAB proteins in MTB was retrieved with the minimum required interaction score of 0.4. Here, the interaction contains both known and predicted PPIs, which are physical and functional interactions with confidence scores. These interactions are the bases to construct the network, so to obtain reliability; we used a medium confidence score. The duplicate interactions were removed from the interaction table and imported to the Cytoscape [19] to visualize the network.
2.2. Analysis of Statistical Parameters of MtrAB-PPI Network
To understand the topological behavior and importance of the key nodes, we used different statistical parameters such as degree distribution (P(k)), clustering coefficient C(k), neighborhood connectivity CN (k), and closeness centrality (Cc) using Network Analyzer a Cytoscape plugin [20].
Degree (K) and (P(k)).
Degree (K) denotes the number of interactions of a particular node with other nodes present in a network. The higher degree of nodes helps to identify the central nodes of the network which were further considered as hubs [21]. It can be determined by (Eq. 1).
 |
Where Nk denotes the total number of nodes having k degree whereas N denotes a node. The behavior of the networks can be categorized as random, small-world, scale-free, and hierarchical. In the small-world and random networks, the maximum number of nodes has a similar degree followed the poison distribution law, whereas, in scale-free networks, fewer nodes have a larger degree and followed power-law distribution P (k)~ k-γ. The γ values define the network nature and mode of organization of nodes. (i) if γ is 2≥ γ ≤3, larger degree nodes hold lesser degree nodes and the larger number of separated distributed nodes together, (ii) if γ ≤ 2, showed the importance of consisted functional modules, a hierarchical nature, and (iii) if γ > 3 no relationship between the nodes losing scale-free topology [22].
Neighborhood connectivity CN (k).
Using the topological property CN (k), we can identify the relative mode of connectivity between interacting neighboring nodes and it can be determined (Eq. 2)
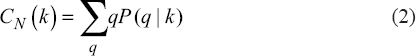 |
where P(q?k) denotes the conditional probability to generate and create connections with nodes consisting of degree (k) to another node with degree q. [23]. For scale-free network topology, the value of CN (k) is constant, whereas CN (k) followed power law distribution, that is, CN (k)~kβ, where, β ~ 0.5 in hierarchical behavior.
Clustering coefficient C(k).
It indicates the extent to which internal connections between a node’s neighborhood can be formed, the strength of connections, and cluster organization. For any single node, it can be determined using the equation C(ki) = 2mi/ki (ki-1) where mi represent all of the links to its nearest neighbors. For scale-free and random networks, the value of C(k) is constant [24] means it is independent of k but in the case of a hierarchical network, it is dependent on a value of C(k)~k-α where α ~ 1 [25].
Closeness centrality (Cc).
It can be determined by calculating the “shortest path lengths” between nodes presented in a network. It is the reciprocal of farness and can be calculated by (Eq.3)
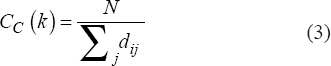 |
where, dij is the sum of all the shortest distance between pair of nodes i and j, whereas N is the nodes present in the network. A node’s (CC) the value indicates how well it can transmit information throughout the network; a low value indicates its capacity to receive information faster.
2.3. Identification of the Nodes Highly Regulating mtrAB Network
According to the hypothesis, proteins with a large number of interacting partners in their network generally serve crucial roles in cells and showed their essentiality. In the constructed network, the proteins with high degrees interact with many other proteins and represented a key regulator of the network. In this study, we used a network analyzer [20] to identify the hub proteins’ communication with many other significant proteins. Removal of hubs (higher degree proteins) may change the structural properties or stability in the network called the centrality-lethality rule [21]. In our analysis, we observed the network organization in the absence of higher degree nodes having large modules interactions one at a time, we considered the betweenness centrality (BC) parameter to re-analyze the rearranged network to measure regulating capacities of key nodes.
2.4. Module Construction and its Association with Hubs
To identify the number of clusters/modules (highly connected nodes or clusters of interactions) in the Mtrab PPIN, we used MCODE [26], a module prediction tool that identified the clusters consist of larger interconnections between nodes. The algorithm uses a three-stage process: (i) Weighting: The nodes with the most linked neighbors receive a higher score. (ii) Molecular complex prediction: Recursively add nodes to the complex that are over a specified threshold, starting with the highest-weighted node (seed). (iii) Post-processing: Filters are applied to increase cluster quality (haircut and fluff). We used default parameter values of MCODE, node score cutoff (0.2), haircut, node density cutoff (0.1), K-score (2), maximum depth (100). To construct high-scoring modules in the network, we used the following default parameters of MCODE Cytoscape plugin. Here, we considered the top five modules that consist highest M-score. The identified hubs showed different interactions with a module, which further considered crucial nodes of the network.
2.5. Gene Ontology Enrichment Analysis of Modules
To understand the biological importance of functional modules, we annotated modules functions using the PANTHER tool. It combines gene-related information, such as gene function and cellular location to identify the gene annotation [27]. This tool provides a comprehensive set of efficient and concise annotations to the various genes and can categorize the terms as molecular functions, biological process (BP), and cellular components (CC).
3. RESULTS AND DISCUSSION
3.1. MtrA and MtrB Proteins in M. tuberculosis Strongly Regulated by Rv1364c
After the removal of duplicity in the genes, we constructed the two key proteins (MtrA and MtrB) in MTB, a based PPI network. Constructed MtrAB-PPIN consisted 909 edges (connections) in between 94 nodes (proteins) [Figure 1]. We found five sparsely distributed nodes namely, Rv1364c, regX3, devS, dosT, and gltB showed a high strength to attract a large number of nodes consists a lesser degree. These five proteins showed a high perturbation degree. Since the network is based on MtrA and MtrB interacting proteins, we found a degree of 73 and 51 for MtrB and MtrA, respectively. The key hub protein apart from these two (MtrA and MtrB) proteins has a degree of 48, 38, 37, 37, and 37 for Rv1364c, regX3, devS, dosT, and gltB, respectively. All five hub proteins were interacting with MtrB, but four of the hub proteins interact with MtrA except regX3.
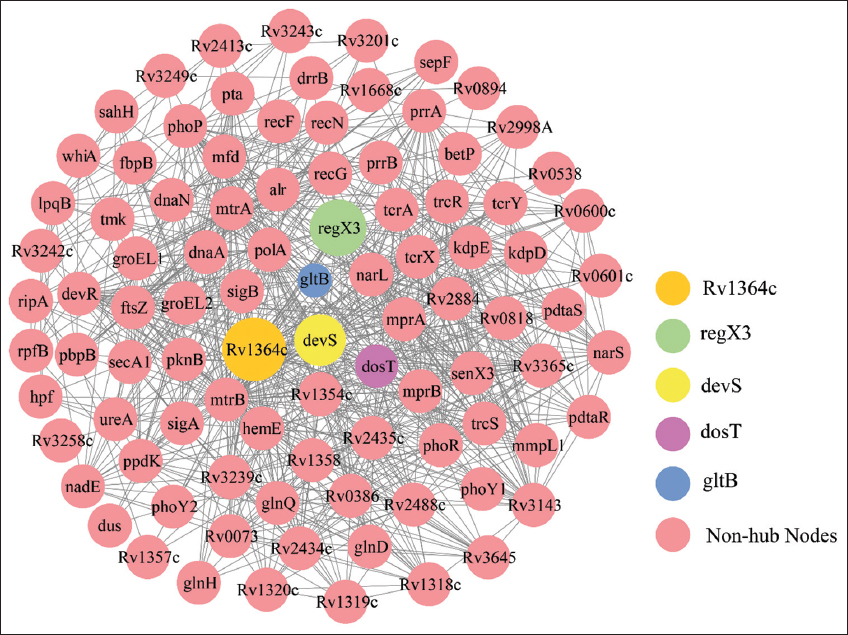 | Figure 1: The hub in the protein-protein interaction of the MtrAB network. Expanded view of the network imported from Cytoscape, where nodes represent proteins and edge the physical interaction. The hub nodes circle size showed according to the degree (larger to smaller). [Click here to view] |
3.2. Statistical and Topological Behavior of the Network
Based on a statistical analysis of the MtrAB-PPIN, we found network followed a power law distribution with degree means fewer nodes having a higher degree but a larger number of nodes having a lesser degree. The negative exponential value in (P(k)) (γ = −0.35, correlation coefficient (r) = 0.37) provides a hierarchical scale-free (presence of modules and sparsely distributed hubs) nature to the network [Figure 2a] [28]. Both CN (k) and C(k) resulted a negative exponent value (β = 53.86 ± 0.23) and (α = 0.96 ± 0.26); with their corresponding r values (r = 0.76) and (r = 0.65) and followed power law distribution [Figure 2b and c]. The exponential values depicted that MtrAB-PPIN consisted of a hierarchical scale-free nature. The MtrAB-PPIN consisted of a disassortative behavior means that hubs play important role in network stability. To, recognize the signal processing strength of the hubs we included closeness centrality (Cc) topological parameter which showed that positive exponents value (η =+0.28) with corresponding (r = 0.90) indicated the leading hubs controlling the network stability with a higher strength [Figure 2d]. The topological and centrality parameters resulted from MtrAB-PPIN following a scale-free nature with a system level of organization (hierarchical scale-free).
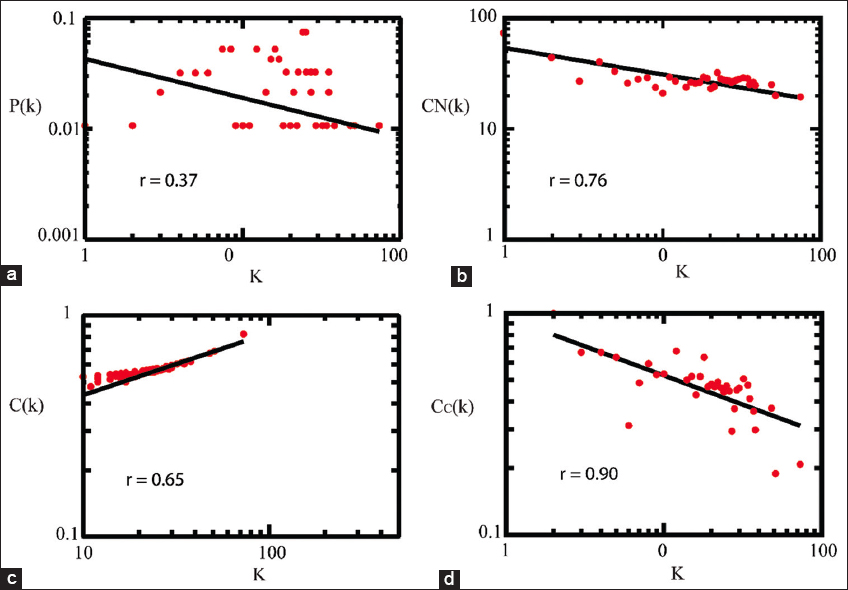 | Figure 2: The topological and centrality properties of the network represented with correlation coefficient values (r). (a) probability of degree distribution P(k), (b) average neighborhood connectivity (CN(k)), (c) average clustering coefficient C(k), (d) closeness centrality (CC). All these properties follow the power law scale. [Click here to view] |
3.3. Nodes Highly Regulating MtrAB Network
To identify the important genes in the network, we performed an independent hub-removal (gene knockout) experiment. Removal of crucial hubs may change the network properties referred to as the centrality-lethality rule [21]. In a hierarchical network, the hubs protein is less important as compared to the functional modules of the network. When it comes to network control, hubs are not as crucial as modules but may involve in the regulatory mechanisms of the network. In general, to understand the importance of a hub node remove the node from the network and then re-analyze the network’s topological properties [29]. Here, we considered the BC parameter which may show a significant change. We removed all 5 hubs once at a time and re-analyzed them by deleting them from the complete PPI network. We found one hub gltB showed significant changes in BC that could increase signal propagation capacity in regulating mtrAB [Table 1]. The removal of the other four hubs (Rv1364c, regX3, dosT, and devS showed a same pattern of the regulation [Figure 3]. Due to the presence of modules that provided hierarchical nature to the network and on hub removal the network stability is still maintained. Most likely, the network’s crosstalk between these hub proteins and functional modules aims to preserve the network’s structural characteristics.
Table 1: Average betweenness centrality values of complete MtrAB-PPIN and after the removal of hubs.
| Name of hub-removed | Average betweenness centrality (BC) |
|---|---|
| Complete network | 0.009258154 |
| devS | 0.009429794 |
| dosT | 0.009419519 |
| gltb* | 0.87840418 |
| reg×3 | 0.009468325 |
| Rv1364c | 0.009465756 |
“*” hub showed significant changes in betweenness centrality.
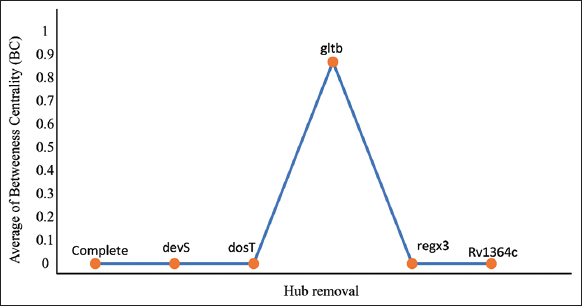 | Figure 3: The representation of the network property comparison of average betweenness centrality (BC) of complete MtrAB-PPIN and after the removal of hubs. [Click here to view] |
3.4. The Modular Structure of the MtrAB Network
The presence of modules in the network provides statistical and functional significance to interacting clusters of nodes, which leads to the formation of a functional community in the network. We identified five such significant modules in the MtrAB network. Here, we identified the top five modules based on the MCODE score (M-score). The module-1 consisted 16 nodes, 62 edges with a M-scoring of 8.26 [Figure 4a]. The module-2, 3, 4, and 5 had 23 nodes (scoring value 7.90) [Figure 4b], eight nodes (scoring value 4.28) [Figure 4c], five nodes (scoring value 3) [Figure 4d], ten nodes (scoring value 2.88) [Figure 4e] with the corresponding edges of 87, 15, 6, and 13, respectively. Among five significant hubs, three hubs present in module-3 and two hubs present in module-2 showed that the majority of the notably large hubs not only controlled their module but also influenced other modules to control network regulation. MtrB protein is present in module-1, and MtrA was not observed in all five modules, which suggested it is indirectly related to modular functionality; with a chance to cross-talk between the module through this protein. We also found modules were linked with sparsely distributed nodes, which provides the possibility of module cross-talk. We identified that these modules communicated with one another through the five most important hubs in the network.
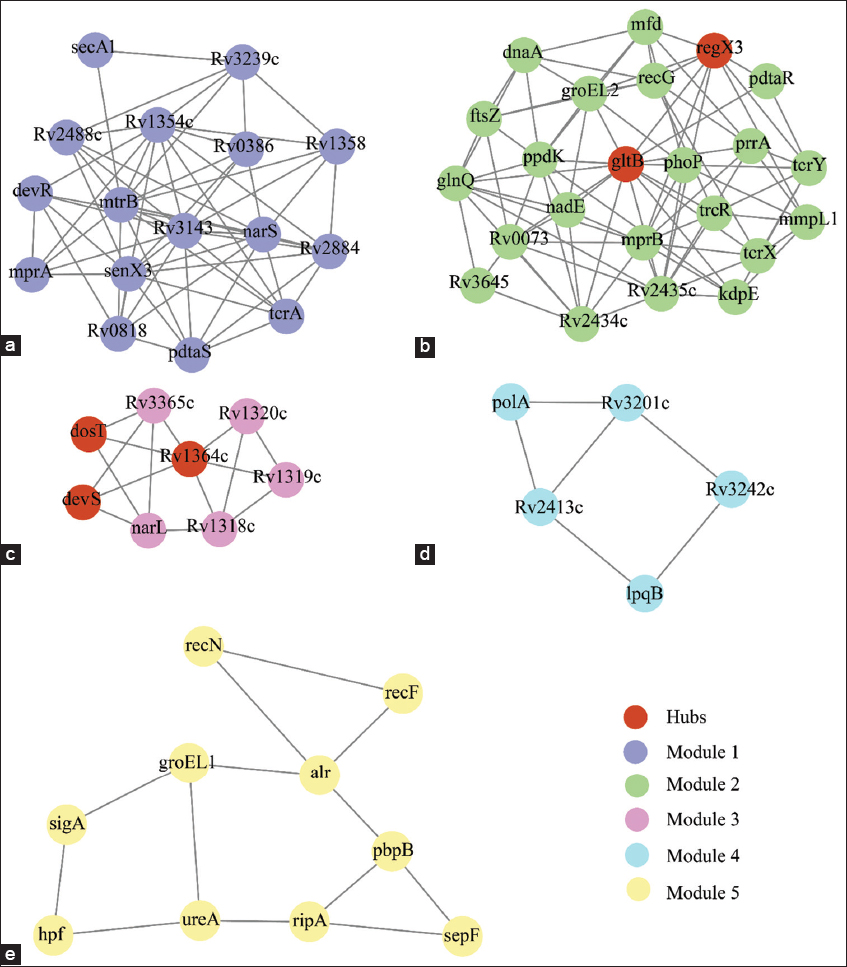 | Figure 4: Structure of the modules in the MtrAB network. MCODE tool used to construct the modules. In the modules 1-5 (a-e) all the nodes are in filled circles with the corresponding edges in lines. [Click here to view] |
3.5. Hubs and Modules Interaction in MtrAB Network Proven Rv1364c and regX3 are Crucial Proteins
The hub protein can directly cross-talk with the nodes present in modules to control the functionality of modules. Rv1364c hub protein showed the maximum interacting strength with five modules, followed by gltB, dosT, devS, and regX3 suggested that hubs were the key mediators of modules [Table 2 and Figure 5]. However, Rv1364c protein is not connected with any of the proteins in module-4. The Rv1364c hub protein interacted with most of the proteins in module-1, 2, and 3. It was identified that increased transcription of a potential sigma factor regulatory gene, Rv1364c, occurs, in Mycobacterium bovis (BCG) consequent to phagocytosis by macrophage [30]. The regX3 showed the fewer interactions with five modules compared to other hubs in the network, but they have a high number of interactions with module-5 compared to other hub proteins. Activated RegX3 limits persister formation during growth under phosphate-limiting conditions [31]. Out of five hubs, two hub proteins did not connect with module-4 and the other three have only one connection each, here MtrB protein interacts with all the proteins in module-4. MtrA is not present in any of the modules, they interact with all the modules with a total connection of 31 [Figure 5]. The increased binding of acetylated MtrA to MtrB is also consistent with MtrA’s repressor action, which also keeps the protein together and available for prompt switching if necessary [32]. We found that Rv1364c regulates one cluster of nodes (module1, 2, and 3), and regX3 hub along with MtrA and MtrB protein regulated module-4 and modules-5. The relationship between hubs and modules suggested that hubs also have capabilities to regulate the functionality of modules, stability, and signal processing in the network. The identified five modules observed in the network provided the hierarchical nature and hub interacting proteins were highlighted. It was clear that these five modules served as the MtrAB network’s primary controller and regulator. The important two hubs (Rv1364c and regX3) in MtrAB network play significant roles through their interaction with two clusters of modules and these two hubs could preserve the stability of the network, information processing. All the five hubs, including MtrB protein present in module-1, 2, and 3 were identified as the most influencing nodes having stronger cross-talk in between the modules that interact with it. MtrA protein, absent in all five modules, but provide a link to cross-talk among the modules and their functions.
Table 2: The total number of nodes and the number of interactions in modules with five hubs.
| Hub interaction networks | Module-1 | Module-2 | Module-3 | Module-4 | Module-5 | Total |
|---|---|---|---|---|---|---|
| devS | 12 | 11 | 3 | 1 | 1 | 28 |
| dosT | 13 | 11 | 3 | 1 | 1 | 29 |
| gltB | 11 | 15 | 1 | 0 | 3 | 30 |
| regX3 | 8 | 9 | 2 | 1 | 4 | 24 |
| Rv1364c | 12 | 17 | 7 | 0 | 2 | 38 |
| Number of nodes | 16 | 23 | 8 | 5 | 10 | 62 |
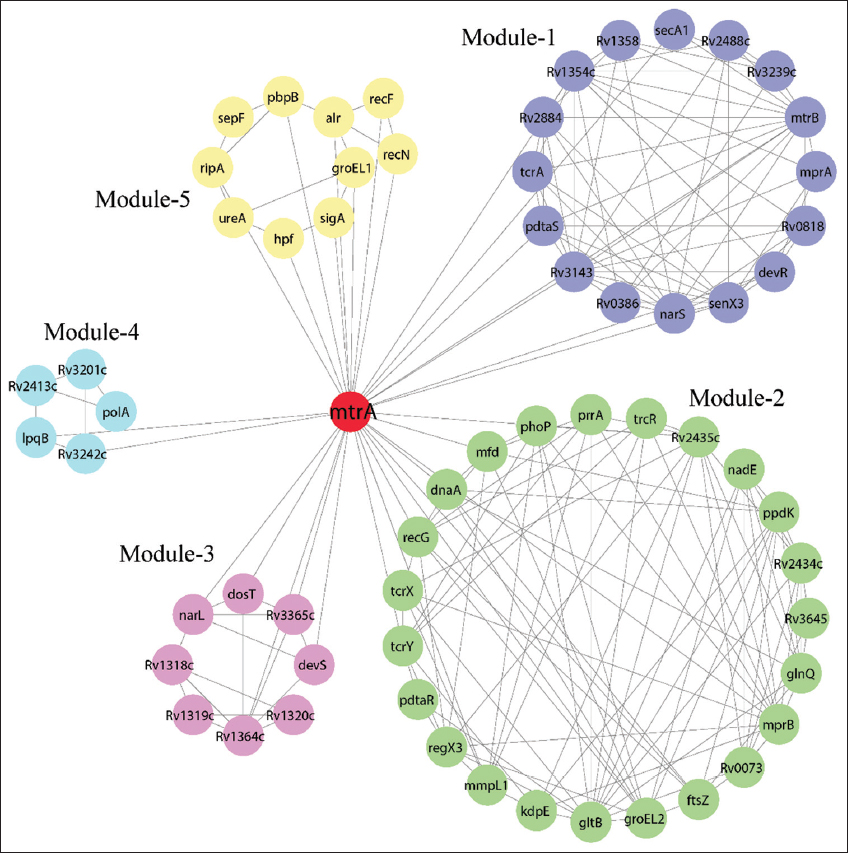 | Figure 5: Cross-talk among the five modules and MtrA hub in the network. MtrA protein interacts with all the modules and hub-interacting nodes were connected via its edges. [Click here to view] |
3.6. Modules Functional Enrichment
We identified that module-1, 2, 3, 4, and 5 were functionally associated with catalytic activity [Figure 6]. Module-4 proteins showed 100% of functional enrichment with catalytic activity. Module-1, 2, and 3 also showed enrichment with molecular transducer activity. Apart from those modules 1 and 2 were functionally related to binding activities. Module-5 showed maximum enrichment of binding function as compared to other modules. All five modules are associated with biological cellular and metabolic processes but mostly with cellular processes. These proteins are mostly found to be localized in a protein complex, cellular anatomical entity, and intracellular. The proteins of module-4 showed 100% ofenrichment as cellular anatomical entities. Module-1 and module-3 proteins showed 50% of enrichment as cellular and another 50% as metabolic processes. Module-2, 4, and 5 also showed an association with response to stimuli BP 6.3%, 4.3%, and 18.2%, respectively [Figure 6].
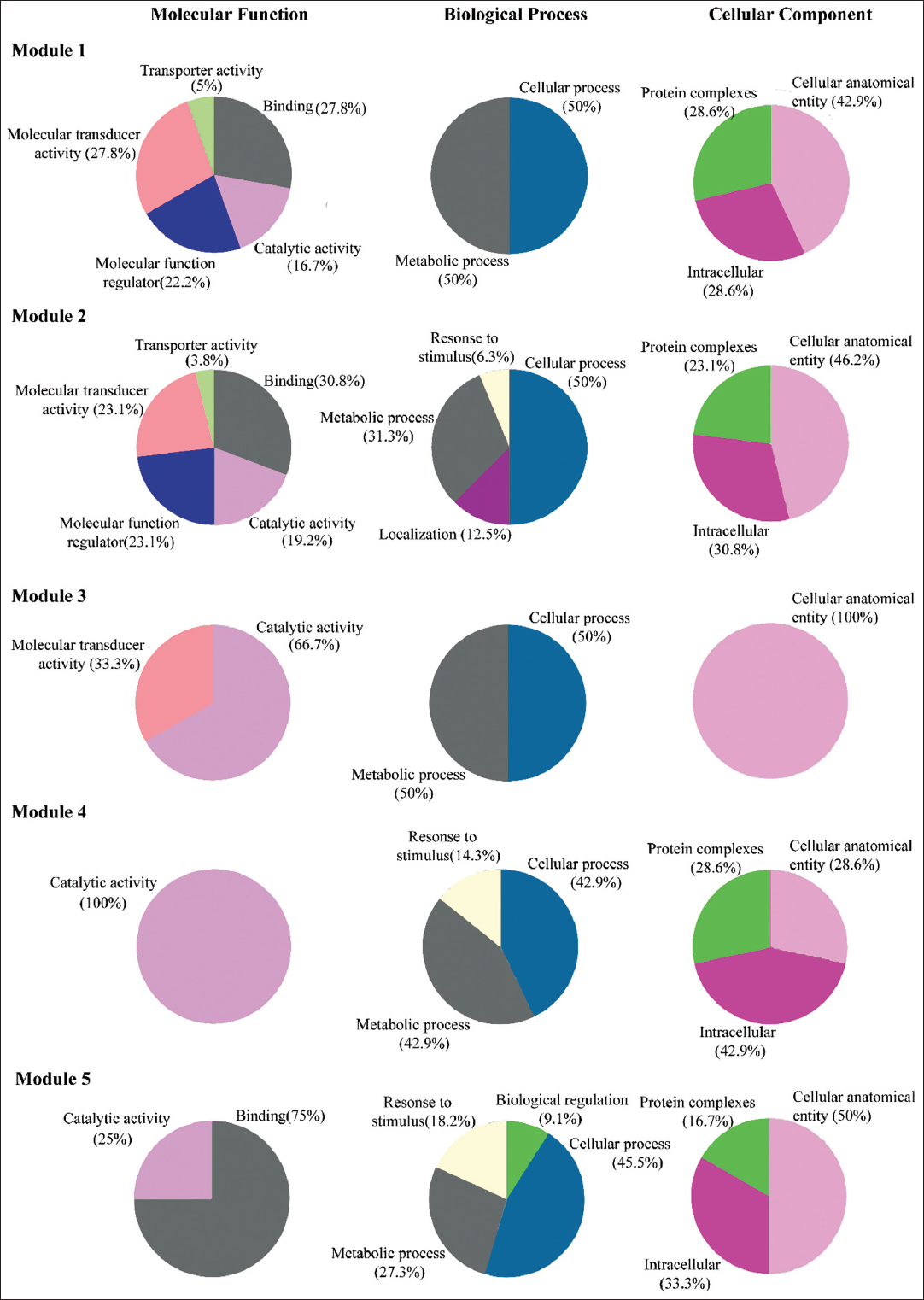 | Figure 6: Functional annotation of five modules. The enrichment analysis in terms of molecular function, biological process, and cellular component shown with percentage represent the function hits of genes. [Click here to view] |
4. DISCUSSION
Network system biology approach helps us to understand the disease biology, such disease pathogenesis, identification of potential biomarkers, drug targets, drug dosages, designing of therapeutic interventions, and synergism discovery [33]. A rapidly expanding area of computational and quantitative research, systems biology integrates a number of disciplines, including microbiome profiling, transcriptomics, proteomics, metabolomics, and genomics. In recent years, systems biology studies have been increasingly implemented in TB research to identify biomarkers and key targets that can showed changes in pathogen viability or its susceptibility to antituberculosis drugs, elucidate host signaling in response to the pathogen, and predict treatment response [4]. Network based systems biology study revealed diseasome and comorbidity associations of systemic sclerosis with different type of cancers [34]. Recent study on congenital heart diseases (CHDs) network regulating heart development and observed that a sub-network also regulates fetal brain development, thereby providing mechanistic insights into the clinical comorbidities between CHDs and neurodevelopmental conditions [35].
In this study, to unravel the signaling challenges associated with MTB, we applied a systems biology approach. Here, we analyzed the constructed PPI network of 94 proteins based on two key proteins (MtrA and MtrB) in MTB. The network analysis results suggested that Rv1364c and regX3 functions as key regulators of the MtrAB network, and both were regulating two modules observed in the network. Out of five key hubs of the network; Rv1364c was found as a major hub. The hub-module interactions showed Rv1364c and regX3 as key proteins, which regulated the network. Among the five hubs, regX3 is the only protein that interacts with other hubs. The connection among Rv1364c, regX3, and MtrB could be a critical three-node motif of the network.
5. CONCLUSION
The modeling and analysis of molecular interaction networks (MINs) could represent valuable resources in defining associated pathways and discovering novel therapeutic targets. Within living organisms, a single protein or other biomolecules will rarely act alone to effect a given function. Instead, there is a complex series of interactions between multiple biomolecules that contributes to a BP. Studying the structure and topology of MINs can help to identify biomolecules that are involved in biological processes and elucidate which biomolecules or processes are dysfunctional in disease.
PPINs have been developed for many different organisms, such as bacteriophages, yeast, bacteria, plants, animals, and human [36]. Recent developments in biomedical research have benefited greatly from PPIN and its uses because it is a useful index to measure their centrality and offer a benchmark for proteins as potential therapeutic targets. Therefore, PPINs become an effective method to understand the complex disease systems like cancer [37], multiple sclerosis [38], and Alzheimer’s disease[39]. A basic aspect of network biology is that proteins implicated in the same disease have a propensity to interact with one another and form disease modules.
To comprehend the etiology of diseases and explain the penetrance and expressivity, it is helpful to study disease modules and provide an idea to the drug target of gene therapy and determines or specifies new disease and drug-protein targets.
6. ACKNOWLEDGMENTS
The authors would like to thank Dr. Kalaiarasan Ponnusamy, Dr. Rita Singh Majumdar, Dr. Malini Rajagopalan, and Dr. Murty Madiraju for evaluable suggestions.
7. AUTHORS’ CONTRIBUTIONS
All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; agreed to submit to the current journal; gave final approval of the version to be published; and agreed to be accountable for all aspects of the work. All the authors are eligible to be an author as per the International Committee of Medical Journal Editors (ICMJE) requirements/guidelines.
8. FUNDING
There is no funding to report.
9. CONFLICTS OF INTEREST
The authors report no financial or any other conflicts of interest in this work.
10. ETHICAL APPROVALS
This study requires no ethical approval as it was based on computational analysis of publicly available data.
11. DATA AVAILABILITY
All the data is available with the authors and shall be provided upon request.
12. PUBLISHER’S NOTE
This journal remains neutral with regard to jurisdictional claims in published institutional affiliation.
REFERENCES
1. Bloom BR, Atun R, Cohen T, Dye C, Fraser H, Gomez GB, et al. Tuberculosis, in major infectious diseases. In:Holmes KK, Bertozzi S, Bloom BR, Jha P, editors. The International Bank for Reconstruction and Development. 3rd ed. Washington, DC:The World Bank;2017.
2. Zumla A, Blasi F, Raviglione M. Rational use of anti-tuberculosis drugs in the eu:Better patient care and less drug resistance. Eur Respir J 2012;39:802-4. [CrossRef]
3. Small MP, Pai M. Tuberculosis diagnosis--time for a game change. N Engl J Med 2010;363:1070-1. [CrossRef]
4. Kontsevaya I, Lange C, Comella-Del-Barrio P, Coarfa C, DiNardo AR, Gillespie SH, et al. Perspectives for systems biology in the management of tuberculosis. Eur Respir Rev 2021;30:200377. [CrossRef]
5. Pankhurst LJ, Elias CD, Votintseva AA, Walker TM, Cole K, Davies J, et al. Rapid, comprehensive, and affordable mycobacterial diagnosis with whole-genome sequencing:A prospective study. Lancet Respir Med 2016;4:49-58. [CrossRef]
6. Consortium CR, Wyllie D, Yang Y, Zhang H, Zhao Y, Zhu B, et al. Prediction of susceptibility to first-line tuberculosis drugs by DNA sequencing. N Engl J Med 2018;379:1403-15. [CrossRef]
7. Strydom N,Gupta SV, Fox WS, Via LE, Bang H, Lee M, et al. Tuberculosis drugs distribution and emergence of resistance in patient's lung lesions:A mechanistic model and tool for regimen and dose optimization. PLoS Med 2019;16:e1002773. [CrossRef]
8. Zschiedrich CP, Keidel V, Szurmant H. Molecular mechanisms of two-component signal transduction. J Mol Biol 2016;428:3752-75. [CrossRef]
9. Rickman L, Saldanha JW, Hunt DM, Hoar DC, Colston MJ, Millar JB, et al. Atwo-component signal transduction system with a PAS domain-containing sensor is required for virulence of Mycobacterium tuberculosis in mice. Biochem Biophys Res Commun2004;314:259-67. [CrossRef]
10. Cole ST, Borch R, Parkhill RB, Garnier T, Churcher C, Harris D, et al. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature 1998;393:537-44. [CrossRef]
11. Snider J, Kotlyar M, Saraon P, Yao Z, Jurisica I, Stagljar I. Fundamentals of protein interaction network mapping. Mol Syst Biol 2015;11:848. [CrossRef]
12. Morgan S, Duguez S, Duddy W. Personalized medicine and molecular interaction networks in amyotrophic lateral sclerosis (ALS):Current knowledge. J Pers Med 2018;8:44. [CrossRef]
13. Mendoza MLZ, Resendis-Antonio O. Modules. Identification methods and biological function. In:Dubitzky W, Wolkenhauer O, Cho KH, Yokota H, editors. Encyclopedia Systems Biology. New York:Springer;2013. 1450-53. [CrossRef]
14. Alcala-Corona SA, Sandoval-Motta S, Espinal-Enriquez J, Hernandez-Lemus E. Modularity in Biological Networks. Front Genet 2021;12:701331. [CrossRef]
15. Kumar R, Haider S. Protein network analysis to prioritize key genes in amyotrophic lateral sclerosis. IBRO Neurosci Rep 2022;12:25-44. [CrossRef]
16. He X, Zhang J. Why do hubs tend to be essential in protein networks?PLoS Genet2006;2:e88. [CrossRef]
17. Maslov S, Sneppen K. Specificity and stability in topology of protein networks. Science 2002;5569:910-13. [CrossRef]
18. Shannon.P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, et al. Cytoscape:A software environment for integrated models of biomolecular interaction networks. Genome Res 2003;13:2498-504. [CrossRef]
19. Assenov Y, Ramirez F, Schelhorn SE, Lengauer T, Albrecht M. Computing topological parameters of biological networks. Bioinformatics 2008;24:282-4. [CrossRef]
20. Jeong H, Mason SP, Barabási AL, Oltvai ZN. Lethality and centrality in protein networks. Nature 2001;411:41-2. [CrossRef]
21. Ravasz E, Barabasi AL. Hierarchical organization in complex networks. Phys Rev E 2003;67:026112. [CrossRef]
22. Traag VA, Krings G, Van Dooren P. Significant scales in community structure. Sci Rep 2013;3:2930. [CrossRef]
23. Barabasi AL, Oltvai ZN. Network biology:Understanding the cell's functional organization. Nat Rev Genet 2004;5:101-13. [CrossRef]
24. Borgatti SP, Everett MG. A Graph-theoretic perspective on centrality. Soc Netw2006;28:466-84. [CrossRef]
25. Bader GD, Hogue CW. An automated method for finding molecular complexes in large protein interaction networks. BMC Bioinformatics 2003;4:2. [CrossRef]
26. Thomas PD, Campbell MJ, Kejariwal A, Mi H, Karlak B, Daverman R, et al. PANTHER:A library of protein families and subfamilies indexed by function. Genome Res 2003;13:2129-41. [CrossRef]
27. Pastor-Satorras R, Vazquez A, Vespignani A. Large scale topological and dynamical properties of the internet. Phys Rev E 2002;65:066130. [CrossRef]
28. Pen X, Wang J, Wang J, Wu FX, Pan Y. Rechecking the centrality-lethality rule in the scope of protein subcellular localization interaction networks. PLoS One 2015;10:e0130743. [CrossRef]
29. Li MS, Waddell SJ, Monahan IM, Mangan JA, Martin SL, Everett MJ, et al. Increased transcription of a potential sigma factor regulatory gene RV1364c in Mycobacterium bovis bcg while residing in macrophages indicates use of alternative promoters. FEMS Microbiol Lett 2004;233:333-9. [CrossRef]
30. Namugenyi SB, Aagesen AM, Elliott SR, Tischler AD. Mycobacterium tuberculosis phoy proteins promote persister formation by mediating pst/senx3-regx3 phosphate sensing. mBio 2017;8:e00494-17. [CrossRef]
31. Singh KK, Athira PJ, Bhardwaj N, Singh DP, Watson U, Saini D. Acetylation of response regulator protein mtra in m. tuberculosis regulates its repressor activity. Front Microbiol 2020;11:516315. [CrossRef]
32. Barabási AL, Silverman EK, Loscalzo J. Network MedicineComplex Systems in Human Disease and Therapeutics. Cambridge, Massachusetts:Harvard University Cambridge Press;2017
33. Islam MK, Rahman MH, Islam MR, Islam MZ, Mamun MM, Azad AK, et al. Network based systems biology approach to identify diseasome and comorbidity associations of systemic sclerosis with cancers. Heliyon 2022;8:e08892. [CrossRef]
34. Wang YJ, Zhang X, Lam CK, Guo H, Wang C, Zhang S, et al. Systems analysis of de novo mutations in congenital heart diseases identified a protein network in the hypoplastic left heart syndrome. Cell Syst 2022;13:895-910.e4 [CrossRef]
35. Hao T, Peng W, Wang Q, Wang B, Sun J. Reconstruction and application of protein-protein interaction network. Int J Mol Sci 2016;17:907. [CrossRef]
36. Pujana MA, Han JD, Starita LM, Stevens KN, Tewari M, Ahn JS, et al. Network modeling links breast cancer susceptibility and centrosome dysfunction. Nat Genet 2007;39:1338-49. [CrossRef]
37. Satoh J, Tabunoki H, Yamamura T, Molecular network of the comprehensive multiple sclerosis brain-lesion proteome. Mult Scler J 2009;15:531-41 [CrossRef]
38. Goni J, Esteban FJ, de Mendizábal NV, Sepulcre J, Ardanza-Trevijano S, Agirrezabal I, et al. Acomputational analysis of protein-protein interaction networks in neurodegenerative diseases. BMC Syst Biol 2008;2:52. [CrossRef]