
1. INTRODUCTION
Rice (Oryza sativa L.) is the most widely consumed staple food of the world’s population belonging to Asia and Africa. Being a semi-aquatic annual plant, rice is highly prone to losses due to various environmental stresses. Many studies had revealed the need for developing varieties tolerant to abiotic and biotic stresses [1]. Methods such as marker assisted breeding, mutation breeding, RNA interference, antisense technology, ZFNs, and TALENs were in use to develop elite traits for abiotic stress tolerance in crops like rice. However, very recently, the, Clustered Regularly Interspaced Short Palindromic Repeats – associated nuclease (CRISPR/Cas) system had come into the limelight as an efficient tool for the development of heritable genetic manipulations in crops [2]. After the successful development of genome-based editing in the model plant Arabidopsis using CRISPR/Cas9 in 2013 [3], numerous agronomic trait-related genes have been edited successfully in various crops. This advancement has paved way for many new possibilities for researchers, including the ability to gain a quicker understanding of the biological plant systems. CRISPR systems are used mainly to achieve enhanced yield efficiency, biofortification, biotic, and abiotic stress tolerance in different crops [1].
Genetic studies in rice have identified OsMADS26 transcription factor as a negative regulator of drought tolerance [4]. OsMADS26 gene is an ortholog of AGL12 and is found to be expressed in roots, shoot, leaf, and panicle throughout all the developmental stages in rice [5,6]. The OsMADS26 overexpression resulted in development of a severe stress phenotype in rice that led to plant death and pigment accumulation. The overexpressed plants showed defective growth, chlorosis, cell death, pigment accumulation, spotted leaves, and senescence [6]. When expressed under the control of a dexamethasone-inducible promoter, differential expression of the genes involved in jasmonic acid biosynthesis as well as regulation of the production of reactive oxygen species was induced [6]. Down-regulation of OsMADS26 was through RNAi showed no dramatic changes in the plant development and the transgenic plants were found to have developed tolerance to drought stress as well as gained resistance toward Magnaporthe oryzae and Xanthomonas oryzae pv oryzae [4]. In these plants, the expression of two drought responsive genes, namely, RAB21 (a rice dehydrin) and SALT STRESS-INDUCED PROTEIN [7] was found to be higher [4,6]. Other stress-related transcription factors were also expressed differentially in accordance with the down regulation of OsMADS26 [8]. In addition to this, it was also identified that the transcript levels of OsMADS26 were higher in older leaves and roots depicting its role in senescence or maturation process [6].
CRISPR/Cas was initially identified as an adaptive mechanism in bacteria for defense against pathogens. In the CRISPR loci, there are repeats that flank the spacer sequence and this, in turn, matches with the sequences of virus, plasmid, or any pathogen genome elements [9]. An A-T rich leader sequence is present upstream of the CRISPR array and it has conserved genes coding for the Cas proteins at the end of the loci [10]. It also includes a single guide-RNA (SgRNA) along with the Cas proteins, which forms the assembly for genome editing. The SgRNA which is customizable is used to guide the Cas endonuclease enzyme to the target site for cleavage [11]. The Protospacer Adjacent Motif (PAM) is present just downstream to the target DNA. Cas9 from Streptomyces pyogenes utilizes “NGG” as the PAM sequence [12]. SgRNA recognizes the PAM sequence and binds to that region in target. The Cas9 protein cleaves the target at region around 3-4 bp upstream of the PAM sequence. The double stranded break in the DNA is repaired by the natural repair mechanisms like Non-homologous End Joining (NHEJ) which is highly error prone and the homology dependent repair (HDR). The error prone repair creates mutations at the target that leads to either activation or knock out of the gene.
The basic protocol for the CRISPR/Cas mediated genome editing has been developed in various crops through cumbersome trials. A protocol for this platform in rice was developed by Shan et al. [13] which shed light for further research in this area. The credibility of this system of editing genome was found more efficient in rice with less off-target effects by carefully studying the pattern, specificity, and heritability of the gene modifications. The results of the study showed that about 11 target genes of two subspecies of rice used for the study inherited the mutations induced through CRISPR/Cas following the Mendelian laws in further generations, reinstating the credibility of using this system for successful and stable genome editing [14].
In the present study, gRNAs were designed and cloned in E. coli to target the OsMADS26 gene in rice. The gRNAs were cloned into the pRGEB32 binary vector [2] and successfully mobilized into Agrobacterium strain EHA105, the protocol of which is outlined here.
2. MATERIALS AND METHODS
2.1. CRISPR/Cas9 Binary Vector
The rice specific CRISPR/Cas9 binary vector pRGEB32 was procured from Addgene, the non-profit plasmid repository (CAT # 63142). The culture was obtained as stab culture. Culture was revived on LB agar medium supplemented with 50.0mg/L Kanamycin. For long-term storage, 15% glycerol stocks were prepared and stored in deep freezers.
2.2. Bacterial Strains
E. coli strain DH5α and Agrobacterium tumefaciens strain EHA105 were used in the study. The bacterial strains used in the study were obtained from DBT-National Institute of Plant Genome Research, New Delhi, India. The cultures were maintained on LB agar plates with suitable antibiotics as well as glycerol stocks.
2.3. Retrieval of Gene Sequence Data of OsMADS26 Gene
The sequence information of the rice transcription factor gene OsMADS26 was obtained from the “Rice Genome Annotation Project” (http://rice.plantbiology.msu.edu). The genomic sequence was saved as FASTA file and used for guide RNA design.
2.4. Designing of Guide RNAs (gRNAs)
The guide RNAs (gRNAs) of 20 bp long, for editing of OsMADS26 gene, were designed using the free online tool CRISPR-P v2.0 (http://crispr.hzau.edu.cn/). Best three-guide RNAs were selected based on the on-score values, GC content, number of off-target sites, and location in the genome. Oligos were synthesized as top and bottom strands of high purity desalted oligos with BsaI restriction sites to facilitate cloning [Table 1].
Table 1: The gRNAs selected for OsMADS26 gene editing.
| S. No. | Guide RNA name | Strand | Sequence (5’- 3’) |
|---|---|---|---|
| 1 | OsMADS26 gRNA 1 (G1) | Top strand | GGAGCTCTCCATCCTCTGCG |
| Bottom strand | CGCAGAGGATGGAGAGCTCC | ||
| 2 | OsMADS26 gRNA 2 (G2) | Top strand | CCGGCCTGCTGAAGAAGGCC |
| Bottom strand | GGCCTTCTTCAGCAGGCCGG | ||
| 3 | OsMADS26 gRNA 3 (G3) | Top strand | ACTAGTTTGGACTAGCTTCG |
| Bottom strand | CGAAGCTAGTCCAAACTAGT |
2.5. Designing of Primers
Primers for amplification of partial gene sequences of OsMADS26 gene (~450 bp) and hygromycin (hpt) resistance gene (1.0 kb) from the vector pRGEB32 were designed manually and analyzed for their features using “Oligoevaluator” tool (Sigma-Aldrich).
2.6. Confirmation of OsMADS26 Gene in Rice Genome
The rice genomic DNA was isolated from the seedlings of Oryza sativa ssp. japonica cultivar Nipponbare following the CTAB method [15]. The target region for CRISPR/Cas9-mediated editing of OsMADS26 gene was amplified using polymerase chain reaction (PCR) using gene specific flanking primers. The PCR reaction and cycling conditions are described in Tables 2 and 3. The PCR product was checked on 1.2 % gel and sequences were confirmed.
Table 2: PCR reaction mixture to confirm OsMADS26 gene target in Nipponbare.
| Reagents | Quantity |
|---|---|
| Rice genomic DNA (50–100 ng) | 2.0 mL |
| 10x buffer with MgCl2 | 5.0 mL |
| dNTP mix (2.0 mM each) | 4.0 mL |
| OsMADS26 FP (10 mM) | 2.0 mL |
| OsMADS26 RP (10 mM) | 2.0 mL |
| Taq DNA Polymerase, GeNei (5U/mL) | 1.0 mL |
| Nuclease free water | 34.0 mL |
| Total reaction volume | 50.0 mL |
Table 3: The PCR program for the amplification of OsMADS26 gene target.
| Initial denaturation | 95°C | 5 min | |
| Denaturation | 95°C | 30 s | 35 cycles |
| Annealing | 60°C | 30 s | |
| Extension | 72°C | 30 s | |
| Final extension | 72°C | 10 min | |
| Hold | 4°C | ∞ |
2.7. CRISPR/Cas9 Vector Construction
For the development of CRISPR/Cas9 editing vector, pRGEB32 plasmid isolated using the Nucleospin® plasmid isolation kit (Macherey-Nagel) was linearized using the restriction enzyme BsaI-HF-v2 enzyme (New England Biolabs, UK). The digestion reaction was set up as shown in Table 4 and incubated in thermal cycler (Agilent, Sure cycler 8800).
Table 4: Reaction set up for vector digestion.
| Reagents | Quantity | Reaction conditions |
|---|---|---|
| pRGEB32 (pDNA) (1.0 mg) | 25.0 mL | • 37C for 2 h |
| Nuclease free water | 19.5 mL | |
| NEB 10x CutSmart buffer | 5.0 mL | |
| NEB BsaI-HF v2 (20U/mL) | 0.5 mL | |
| Total reaction volume | 50.0 mL |
The digested plasmid was analyzed for linearization on 1.0% agarose gel and the vector was purified using Nucleospin® (Macherey-Nagel) PCR clean-up kit. The concentration of plasmid was analyzed using Nanodrop® spectrophotometer (Thermo Scientific, ND-1000). The top and bottom strand oligos of gRNAs were annealed before ligation into pRGEB32 vector. The gRNA ends were phosphorylated using T4 poly nucleotide kinase (PNK) (Thermo Scientific) and used for annealing [Table 5]. After incubation, reaction tubes were kept at room temperature to cool down gradually for around 1 h. The annealed gRNAs were diluted (1:200) before ligation. The digested vector and annealed gRNAs were ligated together and transformed into E. coli strain DH5α [Table 6].
Table 5: The gRNA annealing reaction with T4 PNK.
| Reagents | Quantity | PCR program |
|---|---|---|
| gRNA top strand (100 mM) | 1.0 mL | • 37°C for 60 min |
| gRNA bottom strand (100 mM) | 1.0 mL | |
| 10x T4 DNA ligase buffer | 1.0 mL | |
| T4 PNK, Thermo scientific (10U/mL) | 0.5 mL | |
| Nuclease free water | 6.5 mL | |
| Total reaction volume | 10.0 mL |
Table 6: Ligation reaction for phosphorylated gRNAs with pRGEB32 vector.
| Reagents | Quantity |
|---|---|
| pRGEB32 digested and purified (~50 ng) | 4.0 mL |
| Annealed gRNAs (5.0mM each) | 2.0 mL |
| 10x T4 DNA ligase buffer | 1.0 mL |
| T4 DNA ligase, Thermo Scientific (5U/mL) | 1.0 mL |
| Nuclease free water | 2.0 mL |
| Total reaction volume | 10.0 mL |
2.8. Transformation of Vector Construct into E. coli
The E. coli strain DH5α was transformed with the ligation mixture containing gRNAs ligated in pRGEB32 binary vector. For this, the competent cells of DH5α were prepared and transformed using the “Bacterial Transformation kit GeneSure™” (PureGene life sciences, USA). After transformation, the bacterial cells were plated onto LB agar plates supplemented with Kanamycin (50.0 mg/L) and incubated for 16–18 h at 37oC.
2.9. Confirmation of the CRISPR/Cas9-gRNA Construct in DH5α
The putative positive clones in E. coli were identified by colony PCR using specific primers followed by sequencing of plasmids isolated from the identified positive colonies. Colony PCR was performed using forward primer specific to gRNA cloned and reverse primer specific to vector (Universal M13 Reverse primer). The PCR reaction and program set up are as given in Tables 7 and 8. Sequence analysis was performed using Sequencher sequence analysis software (Gene Codes Corporation, USA).
Table 7: Reaction set up for colony PCR.
| Reagents | Quantity |
|---|---|
| Template colony | 10.0 mL |
| 10x buffer with MgCl2 | 2.5 mL |
| dNTP mix (2.0 mM each) | 2.0 mL |
| Forward primer (10 mM) | 1.0 mL |
| Reverse primer (10 mM) | 1.0 mL |
| Taq DNA polymerase, GeNei (5U/mL) | 0.5 mL |
| Nuclease free water | 8.0 mL |
| Total reaction volume | 25.0 mL |
Table 8: PCR program for colony PCR.
| Initial denaturation | 95°C | 10 min | |
| Denaturation | 95°C | 30 s | 35 cycles |
| Annealing | 58°C | 30 s | |
| Extension | 72°C | 30–60 s | |
| Final extension | 72°C | 10 min | |
| Final hold | 4°C | ∞ |
2.10. Transformation of the Positive Clones into Agrobacterium tumefaciens Strain EHA105 and Confirmation of the CRISPR/Cas9 Constructs in EHA105
The OsMADS26 CRISPR/Cas9 gRNA constructs confirmed in E. coli were further mobilized into Agrobacterium strain EHA105. The competent cells of EHA105 were prepared by calcium chloride method and transformed using freeze-thaw method [16]. For transformation, competent cells were mixed with recombinant plasmids (0.5–1.0 μg pDNA) and incubated on ice for 30 min. The mixture was frozen in liquid nitrogen for 5 min, followed by thawing at 37°C for 5 min. The cells were incubated at 28oC for 4 h in LB broth. After harvesting, the cells were plated onto LB agar plates supplemented with Kanamycin (50.0mg/L) and Rifampicin (20.0 mg/L). The plates were incubated for 48 h at 28°C. The CRISPR/Cas9 constructs were confirmed in EHA105 using colony PCR with hygromycin resistance gene (hpt) specific primers.
3. RESULTS
The CRISPR/Cas9 binary vector pRGEB32 is 15.9 kb in size [2] and contains rice codon optimized Cas9 gene under rice ubiquitin promoter, BsaI restriction site for guide RNA (gRNA) cloning, and selectable markers for both bacteria (Kanamycin) and plant (Hygromycin) selection after genetic transformation. The single guide RNA (sgRNA) cassette is cloned under the control of rice U3 promoter [Figure 1].
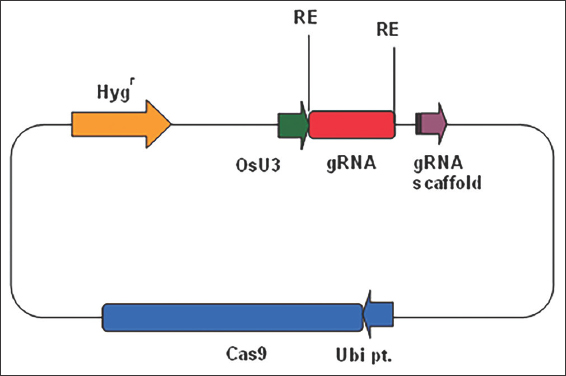 | Figure 1: Schematic representation of the gRNA constructs design for the study. The gRNAs are cloned in between the BsaI sites and expressed under OsU3 promoter. Cas9 is driven by rice ubiquitin promoter. Hygromycin is used as the plant selection marker. [Click here to view] |
3.1. Retrieval of OsMADS26 Gene Sequence
The locus ID of OsMADS26 gene was identified as LOC_Os08g02070 and the gene was found to be located on chromosome 8 of rice genome. The FASTA file of the sequence was downloaded from http://rice.plantbiology.msu.edu for further studies. The genomic sequence of the gene is about 2382 bp and has a CDS (Coding Sequence) length of about 669 bp. The gene contains seven exons and six introns.
3.2. Designing of Guide RNAs (gRNAs)
The gRNAs selected from CRISPR-P v2.0 result window for targeted editing of the OsMADS26 gene are given in Table 1. The three gRNAs of 20 bp length were selected based on their on-score value, off-target sites, GC content, and location in genome. The gRNAs located on the coding sequences (CDS) toward 5’ end of the gene were majorly considered. The first two gRNAs were selected from the CDS region and the third gRNA was selected from the 5’UTR (Untranslated region). The first two gRNAs selected were from the plus strand and the third gRNA was from the minus strand. The restriction sites for the Type IIS restriction enzyme BsaI were added to top and bottom strands at 5’ end to enable cloning into the binary vector.
3.3. Confirmation of OsMADS26 Gene Target Region in Rice
The OsMADS26 gene target sequences in the Nipponbare genome were confirmed by polymerase chain reaction (PCR) using gene specific primers flanking the target region [Figure 2]. The PCR product was eluted and sequenced to check the sequences. The sequencing result was analyzed using BLAST and Clustal Omega programs to confirm the correctness of sequences [Figure 3]. The sequencing result showed 100% similarity of the target region with OsMADS26 gene sequences available in MSU rice genome annotation project database.
 | Figure 2: Gel picture of PCR amplified OsMADS26 gene target (~450 bp), L- 1 kb Plus Ladder, 1- PCR amplified OsMADS26 gene target from rice genome. [Click here to view] |
 | Figure 3: Multiple sequence alignment of OsMADS26 PCR product. Sequence alignment result window of Clustal Omega is shown. Sequence of the PCR product is aligned with OsMADS26 gene sequence available in Rice Genome Annotation Project database (LOC_Os08g02070). [Click here to view] |
3.4. CRISPR/Cas9 Vector Construction and Cloning into E. coli
The BsaI digested vector was electrophoresed on 1% agarose gel to check its linearization [Figure 4]. Digested vector was further ligated with the annealed gRNAs and were used for cloning into E. coli. Bacterial colonies were obtained on the LB agar plates supplemented with suitable antibiotics after overnight incubation following transformation. Each colony was picked up to screen for the presence of inserts using colony PCR with vector and gRNA specific primers [Figure 5]. Clear bands of expected size of ~250 bp were observed on 1.5% agarose gel. The constructs were labeled as OsMADS26 #G1, #G2, and #G3, respectively, based on the gRNA cloned. The PCR positive colonies were selected and used for further plasmid isolation and sequencing. These isolated plasmids were then sequenced using universal M13 reverse primer and the sequencing result was confirmed using sequence alignment tool of Sequencher software [Figure 6a and b]. Colony number 1 and 7 of gRNA-1 construct (pRGEB32:OsMADS26 #G1-1 and pRGEB32:OsMADS26 #G1-7) and colony 3 of gRNA 3 (pRGEB32:OsMADS26 #G3-3) showed insertion of respective gRNAs within the vector backbone. Even after multiple rounds of transformations, positive clones could not be obtained in gRNA-2 and hence not forwarded further.
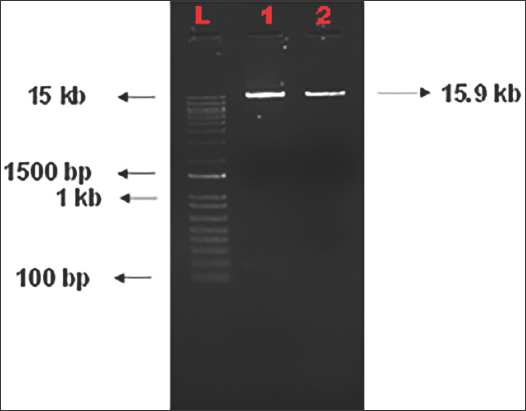 | Figure 4: BsaI restriction digestion profile of the binary vector pRGEB32. L- 1 kb plus ladder, 1- undigested vector and 2- digested vector. [Click here to view] |
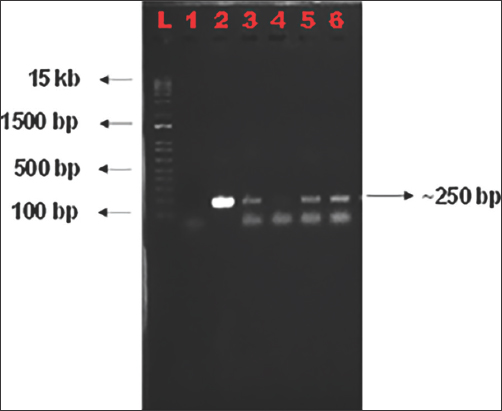 | Figure 5: Colony PCR profile of DH5α colonies. Colony PCR was performed with specific primers to check for the insertion of gRNAs. L- 1 kb Plus Ladder, 1- Non-template control, 2- pRGEB32:OsMADS26 #G3-3, 3- pRGEB32:OsMADS26 #G1-1, 4- pRGEB32: OsMADS26 #G1-4, 5- pRGEB32:OsMADS26 #G1-7, and 6- pRGEB32:OsMADS26 #G1-9. [Click here to view] |
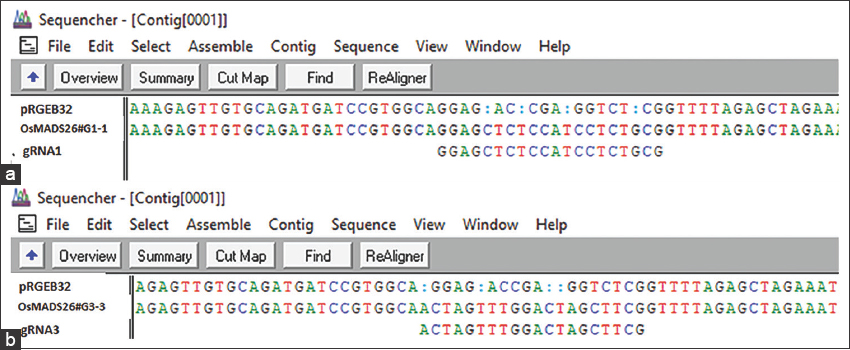 | Figure 6: Sequence alignment file showing the gRNA cloned region in the binary vector. (a) pRGEB32:OsMADS26 #G1-1 construct (b) pRGEB32:OsMADS26 #G3-3 construct. Alignment of the clones with pRGEB32 empty vector and respective gRNA used for cloning is shown. Sequencher sequence analysis software was used to analyze the sequences. [Click here to view] |
3.5. Mobilization of Guide Constructs into Agrobacterium EHA105
The CRISPR/Cas9 gRNA construct for OsMADS26 #G1 and #G3 confirmed as positive clones after sequence analysis was further mobilized into EHA105 strain of A. tumefaciens. The recombinant plasmids, pRGEB32:OsMADS26 #G1-1 and pRGEB32:OsMADS26 #G3-3, were mobilized to EHA105 using freeze-thaw method. After 48 h of incubation at 28°C, 13 and 24 colonies, respectively, were observed on both the transformed plates.
3.6. Confirmation of the CRISPR/Cas9 gRNA Constructs of OsMADS26 in EHA105
The positive clones were confirmed by colony PCR using primers specific to hygromycin resistance gene. Two colonies for OsMADS26 #G1 and one colony for OsMADS26 #G3 were taken for colony PCR. Expected amplicon size of around 1.0 kb was observed in all colonies on 1% agarose gel [Figure 7].
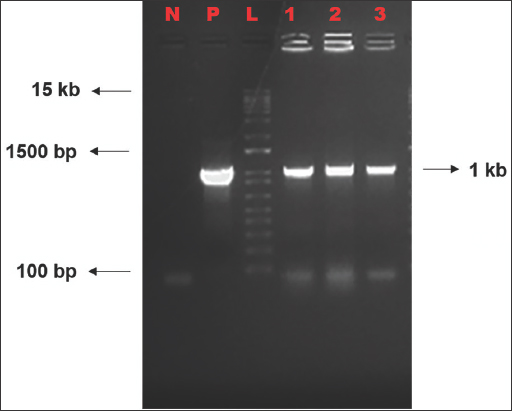 | Figure 7: Colony PCR of colonies obtained in A. tumefaciens, EHA105. N- Non template control, P- positive control (empty pRGEB32), L- 1 kb Plus Ladder, 1- OsMADS26 #G1-1, 2- OsMADS26 #G1-2, and 3- OsMADS26 #G3-1. [Click here to view] |
4. DISCUSSION
The existence of immense genetic variability in the germplasm for drought tolerance in rice has been identified [17]. One of the important transcription factors (TF) family found in rice is MADS-box TF family. About 75 MADS-box genes were identified from rice genome [18]. Transcription factors like OsMADS26 were found to have role in multiple stress responses in rice. The previous studies have demonstrated that the rice transcription factor OsMADS26 is a negative regulator of abiotic and biotic stress tolerance [4].
The pRGEB32 is one of the binary vector systems used for targeted genome editing of rice using the CRISPR/Cas9 system. It was used in the present study because of its reported high efficiency in targeted mutagenesis [2]. The vector system is highly specific to rice. The T-DNA region of the vector has Cas9, guide RNA cloning site and hygromycin resistance genes required for the targeted editing and subsequent selection of the edited events in rice. In pRGEB32, the gRNA cloning site is flanked by BsaI restriction sites. The vector consists of Cas9 from S. pyogenes driven by the rice ubiquitin promoter, a custom polycistronic gRNA assembly (precursor for one or more gRNAs) driven by the rice U3 snoRNA (Pol III) promoter and HPTII (hygromycin resistance) driven by the CaMV35S promoter.
The CRISPR/Cas9 construct was made by ligating the respective gRNAs to the vector pRGEB32 after BsaI digestion. The top and bottom strands of gRNAs were annealed before this. pRGEB32-gRNA construct was then transformed into E. coli strain DH5α. The colonies were observed on the plates after 16–18 h of incubation at 37OC. The transformation into DH5α was really a constraint in the experiment. The larger size of the vector pRGEB32 (15.9 kb) made it difficult to be taken up by the bacterial cells. Hence, the number of colonies observed for both guide RNAs was less. To overcome these constraints, few changes were made during gRNA annealing and ligation reactions.
Very recently, it was reported that while using pRGEB32 for cloning, the transformation efficiency was found to be very low [19]. They developed a protocol specifically for easy transformation of the large sized vectors like pRGEB32 using heat shock method. The study suggests that heat shock temperature of 55OC for 60 s could enhance the transformation efficiency of pRGEB32 compared to the conventional conditions of 42oC for 1 min.
After cloning, the clones observed were confirmed initially by streaking the colonies to fresh plate with antibiotics. Thereafter, colony PCR was done with vector and gRNA specific primers. Positive clones were identified by multiple sequence alignment of the sequences obtained after sequencing. The positive clones obtained in E. coli were further mobilized into A. tumefaciens strain EHA105 [20]. The genetic transformation of EHA105 was carried out by freeze-thaw method [21]. About 13–24 colonies were observed in OsMADS26#G1 and OsMADS26#G3. Even though the method is less efficient compared to the electroporation, it is more adaptable to most laboratory conditions. Agrobacterium-mediated genetic transformation is accepted to be the most reliable method of genetic transformation in rice [22]. Hence, in the present study, the constructs developed will be transformed into rice using Agrobacterium mediated genetic transformation in the future.
5. CONCLUSION
The gRNAs were designed and cloned in to pRGEB32 vector for targeting the rice MADS-box gene OsMADS26 which was found to be a negative regulator of drought tolerance in rice. The CRISPR/Cas9 constructs were successfully mobilized and confirmed in Agrobacterium strain EHA105. The constructs developed can be used to transform rice to develop drought tolerant lines in rice.
6. ACKNOWLEDGMENT
The authors acknowledge the DBT-HRD PG program fellowship to AK and SERB- Women Excellence Award (SERB-WEA/2020/000027) grant to RA. The authors also acknowledge the facilities at Department of Plant Biotechnology, College of Agriculture, KAU, Thrissur, Kerala.
7. AUTHORS’ CONTRIBUTIONS
RA designed experiments, AK conducted experiments, AK and RA interpreted data and wrote the manuscript. All authors read and approved the manuscript.
8. FUNDING
The study was supported by SERB- Women Excellence Award (SERB-WEA/2020/000027) and DBT-HRD PG program.
9. CONFLICTS OF INTEREST
The authors report no financial or any other conflicts of interest in this work.
10. ETHICAL APPROVALS
This study does not involve experiments on animals or human subjects.
11. DATA AVAILABILITY
All data supporting this study are available on request to the corresponding author.
12. PUBLISHER’S NOTE
This journal remains neutral with regard to jurisdictional claims in published institutional affiliation.
REFERENCES
1. Ricroch A, Clairand P, Harwood W. Use of CRISPR systems in plant genome editing:toward new opportunities in agriculture. Emerg Top Life Sci 2017;1:169-82. [CrossRef]
2. Xie K, Minkenberg B, Yang Y. Targeted gene mutation in rice using CRISPR-Cas9 system. Bio Protoc 2014;17:4. [CrossRef]
3. Jiang W, Bikard D, Cox D, Zhang F, Marraffini LA. RNA-guided editing of bacterial genomes using CRISPR-Cas systems. Nat Biotechnol 2013;31:233-9. [CrossRef]
4. Khong G, Pati P, Richaud F, Parizot B, Bidzinski P, Mai C, et al. OsMADS26 negatively regulates resistance to pathogens and drought tolerance in rice. Plant Physiol2015;169:2935-49. [CrossRef]
5. Pelucchi N, Fornara F, Favalli C, Masiero S, Lago C, Colombo L, et al. Comparative analysis of rice MADS-box genes expressed during flower development. Sex Plant Reprod 2002;15:113-22. [CrossRef]
6. Lee S, Woo YM, Ryu SI, Shin YD, Kim WT, Park KY, et al. Further characterization of a rice AGL12 group MADS-box gene, OsMADS26. Plant Physiol 2008;47:156-68. [CrossRef]
7. Claes B, Dekeyser R, Villarroel R, Van den Bulcke M, Bauw G, Van Montagu M, et al. Characterization of a rice gene showing organ-specific expression in response to salt stress and drought. Plant Cell 1990;2:19-27. [CrossRef]
8. Fang YJ, You J, Xie K, Xie WB, Xiong LZ. Systematic sequence analysis and identification of tissue-specific or stress-responsive genes of NAC transcription factor family in rice. Mol Genet Genomics 2008;280:547-63. [CrossRef]
9. Bolotin A, Quinquis B, Sorokin A, Ehrlich SD. Clustered regularly interspaced short palindrome repeats (CRISPRs) have spacers of extrachromosomal origin. Microbiology2005;151:2551-61. [CrossRef]
10. Barman A, Deb B, Chakraborty S. A glance at genome editing with CRISPR–Cas9 technology. Curr Genet 2020;66:447-62. [CrossRef]
11. Jiang F, Doudna JA. CRISPR-Cas9 structures and mechanisms. Annu Rev Biophys2017;46:505-29. [CrossRef]
12. Tahir T, Ali Q, Rashid MS, Malik A. The journey of crispr-cas9 from bacterial defense mechanism to a gene editing tool in both animals and plants. Biol Clin Sci Res J 2020;2020:17. [CrossRef]
13. Shan Q, Wang Y, Li J, Gao C. Genome editing in rice and wheat using the CRISPR/Cas system. Nat Protoc 2014;9:2395-410. [CrossRef]
14. Zhang H, Zhang J, Wei P, Zhang B, Gou F, Feng Z, et al. The CRISPR/Cas9 system produces specific and homozygous targeted gene editing in rice in one generation. Plant Biotechnol J 2014;12:797-807. [CrossRef]
15. Rogers SO, Bendich AJ. Extraction of total cellular DNA from plants, algae and fungi. In:Gelvin SB, Schilperoort RA, editors. Plant Molecular Biology Manual. Belgium:Kluwer Academic Publishers;1994. 183-90. [CrossRef]
16. Holsters M, Waele D, Depicker A, Messens E, Van Montagu M, Schell J. Transfection and transformation of A. tumefaciens. Mol Gen Genet 1978;168:181-7. [CrossRef]
17. Serraj R, McNally K, Slamet-Loedin I, Kohli A, Haefele S, Atlin G, et al. Drought resistance improvement in rice:An integrated genetic and resource management strategy. Plant Prod Sci 2011;14:1-14. [CrossRef]
18. Arora R, Agarwal P, Ray S, Singh AK, Singh VP, Tyagi AK, et al. MADS-box gene family in rice:Genome- wide identification, organization and expression profiling during reproductive development and stress. BMC Genomics 2007;8:242. [CrossRef]
19. Patigu RF, Wijayanti P, Sebastian A, Purwestri YA. Optimization of heat shock temperature and time on the transformation of pRGEB32 into Escherichia coli DH5α. J Biol Trop 2021;21:632-40. [CrossRef]
20. Hood EE, Helmer GL, Fraley RT, Chilton MD. The hypervirulence of Agrobacterium tumefaciens A281 is encoded in a region of pTiBo542 outside of T-DNA. J Bacteriol 1986;168:1291-301. [CrossRef]
21. Weigel D, Glazebrook J. Transformation of agrobacterium using the freeze-thaw method. CSH Protoc 2006;2006:4666. [CrossRef]
22. Hiei Y, Ohta S, Komari T, Komashiro T. Efficient transformation of rice (Oryza sativa L.) mediated by Agrobacterium and sequence analysis of the boundaries of the T-DNA. Plant J 1994;6:271-82. [CrossRef]