
1. INTRODUCTION
Sickle cell anemia (SCA) is an autosomal recessive hemoglobin disorder caused by a mutation in the HBB gene. This mutation leads to substitution of valine for glutamic acid at the 6th amino acid position of the β-globin chain. SCA has been found to be more common in Africa, USA, Mediterranean region, Middle-Eastern countries, and in the Indian subcontinent [1]. In 1952, Lehmann and Cutbush [2] reported the first case of SCA in the Indian subcontinent among the Indian tribal population of Nilgiri hills. In the same year, the case of SCA was reported and documented among the tea garden laborers in upper Assam [3]. SCA is common among the populations of India and a higher prevalence is seen among the ST, SC, and OBC population of Chhattisgarh [4]. The clinical presentation of SCA is very diverse and can be modified by many factors that include age, gender, genetics, hematological, and environmental factors [5,6]. The other factors include fetal hemoglobin (HbF), HBB gene cluster haplotypes, and simultaneous presence of α-thalassemia or glucose-6-phosphate dehydrogenase deficiency [7].
The HBB gene cluster includes epsilon, gamma-G, gamma-A, delta, and beta globin genes. This HBB gene cluster spans approximately 70 kilobases on chromosome 11. Several polymorphic sites flanking the HBB gene cluster have resulted in the delineation of five HBB haplotypes [8,9]. These haplotypes are important for understanding and tracing migration of SCA allele and predicting the severity of SCA disease [10–12]. A better understanding of the HBB haplotypes that influence disease severity is important to predict the clinical outcomes in SCA patients. Hence, the purpose of this study was to elucidate the HBB haplotypes and their correlation with clinical and hematological profile of SCA patients of Chhattisgarh population.
2. MATERIALS AND METHODS
This study was initiated after obtaining approval from the Institutional Ethics Committee of the Sickle Cell Institute Chhattisgarh, Raipur, India. Written informed consent was obtained from study participants. A total of 190 unrelated homozygous sickle cell disease (HbSS) patients confirmed using hemoglobin electrophoresis were included in the study. Values pertaining to complete blood count and hemoglobin high performance liquid chromatography (HPLC) fractions were taken from the patient’s records. All the patients were evaluated for clinical phenotypes such as pain, anemia, jaundice, pneumonia, stroke, and osteonecrosis. Severity scores were determined using the combination of anemia, complications, total leucocyte count (TLC), and transfusion scores. A complete overview of the criteria used for assessing the severity score is given elsewhere [13].
About 3 ml blood samples were collected from each patient. DNA was extracted from whole anticoagulated blood following the standard protocol [14]. DNA concentration and purity were assessed using the nanodrop spectrometer. Nucleotide sequence encompassing important polymorphic sites (5′ to ε, 5′ to Gγ, IVS II Gγ, IVS II Aγ, 5′ψβ, 3′ψβ, 5′β, and IVS II β) in HBB gene cluster were amplified by polymerase chain reaction (PCR) and the fragments were subjected to restricted digestion. The polymorphic sites analyzed; restriction enzymes used, and the consensus sequence of first eight restriction fragment length polymorphism (RFLP) sites for designating five known global HBB haplotypes are documented in Table 1. Based on the presence or absence of a restriction site at the studied polymorphic sites, we assigned haplotypes to all samples. The association of HBB haplotypes with various clinical and hematological variables was analyzed using the analysis of variance. As phase-unknown genotypes were collected, the haplotype sites and frequencies were estimated using maximum likelihood with an expectation-maximization method in Arlequin3.5 software. The descriptive data will be given as mean ± standard deviation (SD). Statistical Package for the Social Sciences software (version 22.0) was used to carry out the statistical analysis of the data.
3. RESULTS
Delineation of haplotypes encompassing the HBB gene cluster in 190 SCA patients revealed four haplotypes such as AI, Senegal, Benin, and Cameroon. Central African (Bantu) haplotype was not observed in our SCA patients. The haplotype frequencies are shown in Figure 1. The AI haplotype is the major haplotype (74.2%), followed by atypical haplotype (21.1%). Senegal, Benin, and Cameroon were found, respectively, in 2.6%, 1.1%, and 1.1% of SCA patients. The atypical haplotypes might have been generated by single and double crossing-over between AI haplotype and ancestral HBB haplotype. The mean age of SCA patients among different HBB haplotype groups is not statically different (p = 0.949) (Table 2). The comparison of mean body mass index (BMI) among haplotypes groups did not reveal significant differences (p = 0.697) (Table 2). Similarly, the mean number of blood transfusions is not significantly different among the haplotype groups (p = 0.160). But the mean number of hospitalizations among haplotype groups is statistically different (p = 0.160). The mean number of hospitalizations in AI and Benin haplotypes is relatively lesser than in other haplotypes (Table 2).
The distribution of different clinical phenotypes among various HBB haplotypes is shown in Figure 2. Distribution of the hematological variables among various HBB haplotypes is documented in Table 3. The differences in the number of red blood cell (RBC), tHb, and HbF levels and other Red cell indices such as mean corpuscular volume (MCV), mean corpuscular hemoglobin (MCH), hematocrit (HCT), and red blood cell distribution width-coefficient variation (RDW-CV) are not statistically significant between different haplotypes (Table 3). The mean HbF levels in the AI Haplotype in this study is 20.0% ± 6.6%, which is slightly higher than the Senegal (17.9 ± 10.5), Benin (13.5 ± 5.0), and Cameroon (13.1 ± 0.9) haplotypes. Levels of total bilirubin, direct bilirubin, serum glutamic oxaloacetic transaminase (SGOT), serum glutamic pyruvic transaminase (SGPT), urea, and creatinine in serum did not differ significantly between the different HBB haplotypes (Table 4). Distribution of different HBB haplotypes in SCA patients with various degrees of severity is shown in Table 5.
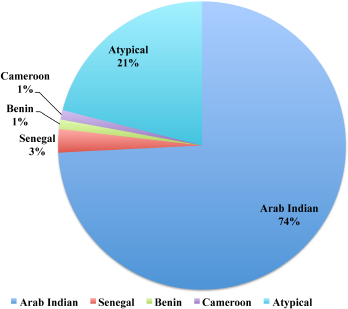 | Figure 1: Distribution of HBB haplotypes among study subjects. [Click here to view] |
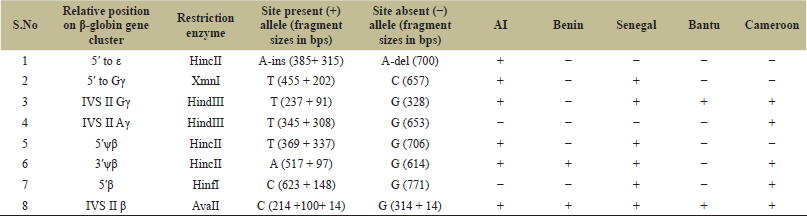 | Table 1: Polymorphic sites analyzed to delineate classical βS haplotypes. [Click here to view] |
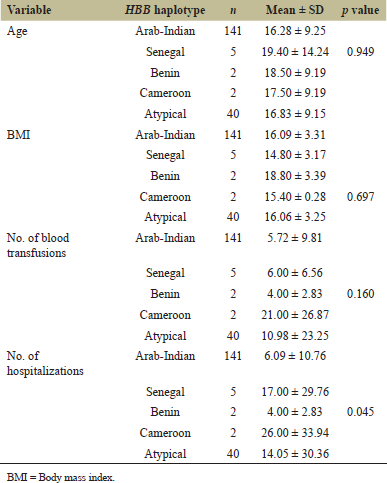 | Table 2: Baseline characteristics of study subjects in each HBB haplotype. [Click here to view] |
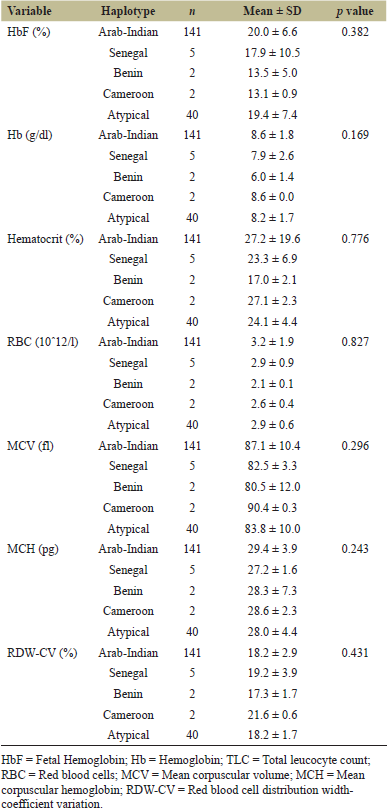 | Table 3: Red blood cells (RBCs) count and blood indices (Hb, HbF, MCH, MCV, and RDW) among various haplotypes. [Click here to view] |
 | Figure 2: Prevalence of clinical complications in SCA patients with various haplotypes. [Click here to view] |
 | Table 4: Levels of total bilirubin, direct bilirubin, SGOT, SGPT, urea, and creatinine in serum among study subjects with various HBB haplotypes. [Click here to view] |
 | Table 5: Prevalence of HBB haplotypes in SCA patient with various degrees of severity. [Click here to view] |
4. DISCUSSION
Analysis of HBB gene cluster haplotypes among SCA patients from the Chhattisgarh region demonstrated presence of AI, Senegal, Benin, and Cameroon haplotypes. Furthermore, several atypical haplotypes were found in this region. Hemoglobin fractions, hematological variables, and clinical complications were compared among haplotypes. To the best of our knowledge, this study is a major study involving 190 well-characterized HbSS patients. Of the 190 HbSS patients analyzed, 74% showed AI haplotype, followed by Senegal, Benin, and Cameroon with the frequencies of 3%, 1%, and 1% respectively. 21% of the chromosomes are of atypical haplotypes (slightly deviate from known haplotypes). Comparison of various biochemical and hematological variables and clinical complications among various haplotype did not reveal significant differences.
HBB haplotypes that are often distinctly associated with a particular geographical region are suggesting that the mutant HBB gene arises separately in these locations [15]. The presence of African-specific HBB haplotypes in Indian populations was documented in many studies [16–18]. Atypical HBB haplotypes that were observed in higher frequencies might have been generated by one of these underlying genetic mechanisms: (a) changes in nucleotides within the restriction site; (b) single and double crossing-over between a typical HBB haplotype and an ancestral HBB haplotype; and (c) non-reciprocal recombination between homologous DNA sequences. Atypical haplotypes are reported in many studies [18–23]. The presence of higher proportions of atypical haplotypes that observed besides AI haplotype in Relli and Thurpu Kapu populations in Andhra Pradesh is probably due to the result of gene conversion [22].
Previous studies that documented the AI haplotype in the Indian subcontinent showed that this haplotype is clinically and hematologically different from other haplotypes and might have evolved differently [15,18]. In consistency with the previous studies, the mean HbF level of the AI haplotype is higher than the other African-specific HBB haplotypes [24–26]. Although the AI haplotype is linked with a high HbF and milder clinical presentations, vasoocclusive episodes still occur among these patients [27,28]. The mean hemoglobin level in the study population is 8.6 g/dl, consistent with the previous studies [29,30]. Increased SGOT/AST levels in the present study (47.42 ± 33.43 U/l) are in consensus with the increased frequency of jaundice (32.05%) and hepatomegaly (6%) occurred in these patients. The increase in total bilirubin levels found in patients with SCA in the present study is corroborated by a previous study in which they found mean values of total bilirubin of 4.85 ± 4.58 mg/dl [30]. The frequency of occurrence of hepatomegaly in the study population is 6%. The striking feature among those patients with hepatomegaly in this study group is that they are associated with an increased aspartate aminotransferase (AST), alanine aminotransferase (ALT), and total bilirubin [31] and may be a useful indicator of severity in sickle cell anemia [32]. The frequency of occurrence of splenomegaly in the study population is 10%. Splenomegaly is one of the main clinical findings among the AI haplotype [18].
In summary, the haplotype analysis of 190 homozygous SCA patients revealed the presence of several known HBB haplotypes. The AI haplotype was the major haplotype, followed by several atypical haplotypes. African haplotypes were found in a lesser frequency. Although our study includes more SCA patients compared to other previous studies, considering the Indian population genetic structure and diversity, our results should be considered as introductory, and our study can serve as a possible tool for future studies. Additional studies based on long-read haplotypes and statistically phased haplotypes in well-characterized SCA patients are warranted.
5. AUTHOR CONTRIBUTIONS
All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; agreed to submit to the current journal; gave final approval of the version to be published; and agree to be accountable for all aspects of the work. All the authors are eligible to be an author as per the international committee of medical journal editors (ICMJE) requirements/guidelines.
6. ETHICAL APPROVALS
This study was initiated after obtaining approval from the Institutional Ethics Committee of the Sickle Cell Institute Chhattisgarh, Raipur, India.
7. CONFLICT OF INTEREST
The authors have no conflicts of interest to declare.
8. FUNDING
This work was supported by Chhattisgarh Council of Science and Technology (CCOST) project (No.2740/CCOST/MRP/2015).
REFERENCES
1. Bhaskar LVKS, Patra PK. Sickle cell disease is autochthonous and unique in Indian populations. Indian J Phys Anthropol Hum Genet 2015;34(2):201–10.
2. Lehmann H, Cutbush M. Sickle-cell trait in southern India. Br Med J 1952;1(4755):404–5. CrossRef
3. Dunlop KJ, Mozumder UK. The occurrence of sickle cell anaemia among a group of tea garden labourers in Upper Assam. Indian Med Gaz 1952;87(9):387–91.
4. Kate SL, Lingojwar DP. Epidemiology of sickle cell disorder in the State of Maharashtra. Int J Hum Genet 2002;2(3):161–7. CrossRef
5. Adekile AD. Limitations of Hb F as a phenotypic modifier in sickle cell disease: study of Kuwaiti Arab patients. Hemoglobin 2011;35(5–6):607–17. CrossRef
6. Embury SH. Alpha thalassemia. A modifier of sickle cell disease. Ann N Y Acad Sci 1989;565:213–21. CrossRef
7. Fong C, Lizarralde-Iragorri MA, Rojas-Gallardo D, Barreto G. Frequency and origin of haplotypes associated with the beta-globin gene cluster in individuals with trait and sickle cell anemia in the Atlantic and Pacific coastal regions of Colombia. Genet Mol Biol 2013;36(4):494–7. CrossRef
8. Powars D, Hiti A. Sickle cell anemia. Beta s gene cluster haplotypes as genetic markers for severe disease expression. Am J Dis Child 1993;147(11):1197–202. CrossRef
9. Powars DR, Meiselman HJ, Fisher TC, Hiti A, Johnson C. Beta-S gene cluster haplotypes modulate hematologic and hemorheologic expression in sickle cell anemia. Use in predicting clinical severity. Am J Pediatr Hematol Oncol 1994;16(1):55–61.
10. Pagnier J, Mears JG, Dunda-Belkhodja O, Schaefer-Rego KE, Beldjord C, Nagel RL, et al. Evidence for the multicentric origin of the sickle cell hemoglobin gene in Africa. Proc Natl Acad Sci U S A 1984;81(6):1771–3. CrossRef
11. Serjeant GR. The geography of sickle cell disease: opportunities for understanding its diversity. Ann Saudi Med 1994;14(3):237–46. CrossRef
12. Makani J, Williams TN, Marsh K. Sickle cell disease in Africa: burden and research priorities. Ann Trop Med Parasitol 2007;101(1):3–14. CrossRef
13. van den Tweel XW, van der Lee JH, Howard J, Fijnvandraat K. Measurement of disease severity in patients with sickle cell disease: a systematic review. American Society of Hematology, Washington, DC, 2007. CrossRef
14. Sambrook J, Russell DW. Molecular cloning: a laboratory manual. 3rd edition, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, 2001.
15. Oner C, Dimovski AJ, Olivieri NF, Schiliro G, Codrington JF, Fattoum S, et al. Beta S haplotypes in various world populations. Hum Genet 1992;89(1):99–104. CrossRef
16. Labie D, Srinivas R, Dunda-Belkodja O et al. Haplotypes in tribal Indians bearing the sickle gene: evidence for the unicentric origin of the beta S mutation and the unicentric origin of the tribal populations of India. Hum Biol 1989;61:479–91.
17. Bhagat S, Patra PK, Thakur AS. Fetal haemoglobin and beta-globin gene cluster haplotypes among sickle cell patients in Chhattisgarh. J Clin Diagn Res 2013;7(2):269–72. CrossRef
18. Niranjan Y, Chandak GR, Veerraju P, Singh L. Some atypical and rare sickle cell gene haplotypes in populations of Andhra Pradesh, India. Hum Biol 1999;71(3):333–40.
19. Uma Mahesh KSS, Aggarwal A, Vijaya Bhasker M, Mukhopadhyay R, Saraswathy KN. Distribution pattern of HbS and b-globin gene haplotypes among Koya Dora Tribe of Andhra Pradesh. Int J Hum Genet 2011;11(2):123–6. CrossRef
20. Sarnaik SA, Ballas SK. Molecular characteristics of pediatric patients with sickle cell anemia and stroke. Am J Hematol 2001;67(3):179–82. CrossRef
21. Gonçalves MS, Braga JAP, Gesteira MF, Carvalho MH, Saad STO, Figueiredo MS. Clinical, hematological, and molecular characterization of sickle cell anemia pediatric patients from two different cities in Brazil. Cad Saúde Pública 2005;21(4):1287–90. CrossRef
22. Ramana GV, Chandak GR, Singh L. Sickle cell gene haplotypes in Relli and Thurpu Kapu populations of Andhra Pradesh. Hum Biol 2000;72(3):535–40.
23. Nongbri SRL, Verma HK, Lakkakula B, Patra PK. Presence of atypical beta globin (HBB) gene cluster haplotypes in sickle cell anemia patients of India. Rev Bras Hematol Hemoter 2017;39(2):180–2. CrossRef
24. Nagel RL, Fabry ME, Pagnier J, Zohoun I, Wajcman H, Baudin V, et al. Hematologically and genetically distinct forms of sickle cell anemia in Africa. The Senegal type and the Benin type. N Engl J Med 1985;312(14):880–4. CrossRef
25. Nagel RL, Rao SK, Dunda-Belkhodja O, Connolly MM, Fabry ME, Georges A, et al. The hematologic characteristics of sickle cell anemia bearing the Bantu haplotype: the relationship between G gamma HbF level. Blood 1987;69:1026–30. CrossRef
26. Padmos MA, Roberts GT, Sackey K, Kulozik A, Bail S, Morris JS, et al. Two different forms of homozygous sickle cell disease occur in Saudi Arabia. Br J Haematol 1991;79(1):93–8. CrossRef
27. El-Hazmi MA, Warsy AS, Al-Momen A, Harakati M. Hydroxyurea for the treatment of sickle cell disease. Acta Haematol 1992;88(4):170–4. CrossRef
28. Adekile AD, Haider MZ. Morbidity, beta S haplotype and alpha-globin gene patterns among sickle cell anemia patients in Kuwait. Acta Haematol 1996;96(3):150–4. CrossRef
29. El-Hazmi MA, Warsy AS, Bashir N, Beshlawi A, Hussain IR, Temtamy S, et al. Haplotypes of the beta-globin gene as prognostic factors in sickle-cell disease. East Mediterr Health J 1999;5(6):1154–8.
30. Keikhaei B, Galehdari H, Pedram M, Jaseb K, Bashirpour S, Zandian K, et al. Beta-globin gene cluster haplotypes in Iranian sickle cell patients: relation to some hematologic. Iranian J Blood Cancer 2012;4(3):105–10.
31. Oparinde DP, Oghagbon EK, Okesina AB, Olatunji PO, Ojuawo AO. Role of hepatic enzymes in the biochemical assessment of the severity of sickle cell anemia. Trop Gastroenterol 2006;27(3):118–21.
32. Olatunji PO, Falusi AG. Persistent hepatomegaly: an index of severity in sickle cell anaemia. East Afr Med J 1994;71(11):742–4.