
1. INTRODUCTION
The utilization of engineered cell lines presents significant advantages for both academic and industrial research applications. Engineered cell lines offer significant research advantages by providing a consistent genetic background, ensuring reproducible results, and simplifying experimental design. They enable the exploration of gene activity and biological processes through specific genetic alterations, such as fluorescent markers. These cell lines are crucial for high-throughput drug screening, validating therapeutic targets, and disease modeling, including personalized medicine. Tools like clustered regularly interspaced short palindromic repeats (CRISPR)/Cas9 facilitate long-term studies and precise genetic modifications. Engineered cell lines have lower variability than primary cells, are easier to maintain, and can sometimes replace animal models, addressing ethical concerns. They are also valuable in synthetic biology for developing novel biological functions, providing a controlled and reliable environment for studying complex biological systems. Gene targeting, a common technique, involves the strategic placement of homologous regions flanking transgenic elements via a “donor” plasmid. This configuration facilitates selective integration through the exploitation of endogenous cellular homology-directed repair (HDR) mechanisms. A variety of recombinant proteins, including zinc finger nucleases and transcription activator-like effector nucleases, have been employed to instigate double-strand breaks (DSBs) at designated genomic sites. This action triggers the activation of the endogenous cellular DNA damage response and subsequent repair pathways [1–4]. The precision of gene targeting has been significantly enhanced by the advent of CRISPR-Cas9 technology [5]. The non-homologous end-joining of DSBs can result in mutagenic insertions and deletions, collectively referred to as “Indels.” This process substantially amplifies the frequency of gene targeting events, even as donor-mediated HDR accurately reconstructs the genomic sequence [6]. Consequently, nuclease systems prove instrumental for transgenic integration.
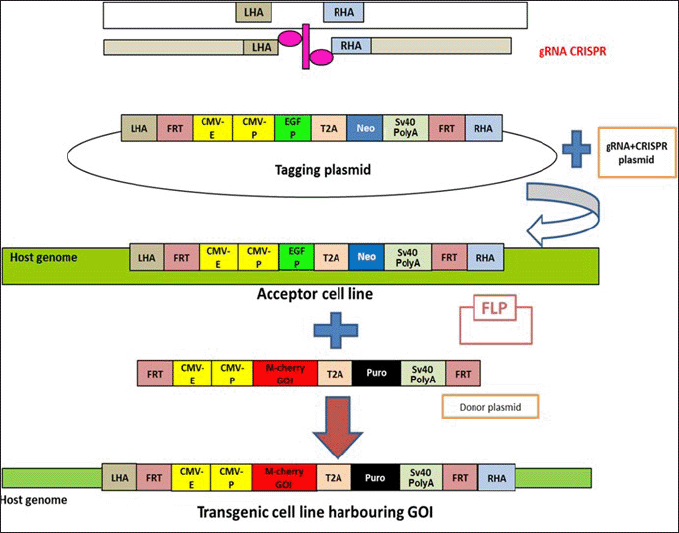
We delineate a methodology for the precise integration of exogenous DNA into predetermined genomic loci in cultured cells. This approach leverages a Cas9-induced DSB to introduce a promoter and flippase recognition target (FRT) site into a specified region. Subsequently, an FRT recombination event facilitates the insertion of a gene of interest (GOI) into the predetermined genomic sequence. An antibiotic resistance gene, expressed from a promoter and incorporated into the genome during the initial phase, ensures the selection of correct integrants. Ultimately, a flippase (Flp)-FRT recombination event excises the selection cassette, leaving behind a precisely targeted GOI.
The Flp-FRT system, a widely employed technique for site-directed recombination technology, originates from Saccharomyces cerevisiae. Utilizing two FRT sites, the Flp recombinase catalyses DNA rearrangements. When constructing vectors containing FRT sites, the directionality must be meticulously considered. Sequence inversion results from opposite orientation, while sequence deletion results from recombination between two FRT site pairs in the same direction. The guide RNAs (gRNA) determines the site of DNA cleavage, and the sequence of the homology arms facilitates homologous recombination (HR). These two factors collectively ascertain the appropriate integration of a GOI into the selected genomic position.
The cell lines utilized to generate transgenic cell lines are indispensable in both industrial and medical research. These cell lines were employed throughout all stages of the cytotoxicity test to validate a drug’s dose response. The creation of a transgenic cell line from scratch is a time-consuming process. However, we have successfully generated transgenic cell lines with minimal off-target effects using CRISPR technology. It is feasible to develop a stable cell line with positive selection markers such as puromycin, G418/Geneticin, Zeocin, and Blasticidin resistance genes. Selection markers can be introduced either on the same plasmid (in cis) with the GOI or on a separate plasmid (in trans), which must be co-transfected with the GOI plasmid. The cis strategy is typically simpler and more likely to yield stable transfectants that express the desired gene and exhibit drug resistance.
Therefore, we have developed a platform that enables researchers to transfect a cell line with the GOI through a single transfection. This platform aims to reduce the time and labor-intensive research required to obtain transgenic cell lines while ensuring high-quality data and minimal off-target effects.
2. MATERIALS AND METHODS
2.1. Cell Lines Used for Making Transgenic Cell-Line
Important cell lines useful for various biotechnological applications such as vaccine production, drug cytotoxicity, metabolism testing, antibody production, gene function research, artificial tissue synthesis, and therapeutic protein synthesis were chosen.
1.1. HEK-293 cell line has epithelial morphology and was obtained from a human embryo kidney. It is employed in toxicity and industrial biotechnology research. It is easy to grow and maintain, with good reproducibility. Additionally, it can be readily transfected for the production of heterogeneous proteins.
1.2 Jurkat cell line was generated from the peripheral blood affected by acute T-cell leukemia. This cell line is employed in immunology and immuno-oncology research.
1.3 Caco-2 is colon epithelial cells obtained from colorectal cancer. It is a good transfection host and is used in cancer and toxicity studies.
1.4 HepG2 is an epithelial-like cell line derived from patients with liver cancer. It is a good transfection host with Insulin and insulin-like growth factor II as an expression marker.
1.5 MCF7 are epithelial cells obtained from metastatic breast tissue. These cells can be used in the study of breast cancer.
1.6 HT-1080 are human epithelial cells that can be utilized in research, and they are obtained from connective tissue from a patient with fibrosarcoma.
1.7 HeLa is a human cervical cancer cell line and has been used in virology, cancer, and so on. Furthermore, HeLa cells are invaluable for space microbiology due to their immortality, allowing indefinite cell division and long-term studies. Their human origin makes them a relevant model for studying space effects on human cells. Despite being cancerous, they maintain genetic stability and have established culturing protocols. HeLa cells respond to environmental stresses like radiation and microgravity, providing insights into space conditions’ effects on human cells. Their standardized nature ensures consistent comparisons across studies, making them essential for space research.
2.2. Adeno-Associated Virus Integration Site 1 (AAVS1) Region & Homology Arm and gRNA Designing and Cloning
Primers were specifically designed for the AAVS1 region, a sequence common across numerous mammalian cell lines (Supplementary Fig. S1). Polymerase chain reaction (PCR) was conducted on the cell lines Hela, HEK293, HepG2, MCF-7, HT1080, CaCO2, and Jurkat (Supplementary Fig. S2). All cell lines were amplified using the AAVS1.Reg.1.F and AAVS1.Reg.1.R primers. Sanger sequencing was employed to identify sequences exhibiting homology across all the cell lines. Based on these identified sequences, five single-guide RNAs (sgRNAs) were designed utilizing the E-CRISP software (Supplementary Fig. S3) [7]. The AAVS1 locus is situated on chromosome 19 and has been documented as a “safe harbour” locus for gene insertion [8]. The region spanning from 981 bp to 1920 bp, as per the NCBI reference sequence: NC_000019.10 (Homo sapiens chromosome 19, GRCh38.p2 Primary Assembly), was amplified. This region is referred to as “AAVS1R1.” The amplification of AAVS1R1 from HEK293 cells was achieved using the following primers: AAVS1R1.F: 5′- TGGCTTCTGCGCCGCCTCTGG-3′ and AAVS1R1.R: 5′- AGGTGGGGGTTAGACCCAAT -3′. The AAVS1R1 fragment was amplified from HEK293 genomic DNA (gDNA) utilizing DreamTaq (Thermo Scientific, USA) and subsequently cloned into the vector pTZ57R using the InsTAclone PCR cloning kit (Thermo Scientific, USA).
Five gRNAs were cloned into the pSpCas9 (BB)-2A-Puro (PX459) construct from Addgene, USA. This construct expresses the gRNA in conjunction with the tracer RNA and Cas9 (hspCas9), using the puromycin resistance gene as a marker. The gRNA target sequences were synthesized as DNA oligonucleotides, with BbsI restriction sites flanking the target sequence, and cloned into the PX459 vector. The top and bottom strand orientations of the guide oligos were annealed in a thermocycler using the following parameters: 37°C for 30 minutes, 95°C for 5 minutes, and a ramp down to 16°C at a rate of 2°C per minute. These were then ligated into the BbsI sites in PX459 and transformed into DH5α competent cells [9]. All the sequences for gRNAs and primers are depicted in Supplementary Table 1.
2.3. Surveyor Nuclease or T7 Endonuclease 1 (T7E1)
All cell lines at 104 cells in 10 ml (105 cells) underwent transfection with 1 μl of Px459-gRNA clones (at 10 ng/ml). Post-transfection (48 hours later), the PX459-gRNA transfected cell lines were subjected to Puromycin selection. After 8 days, viable colonies were pooled for gRNA screening. The “Surveyor assay” based kit was utilized to check for host DNA cleavage. As per the instructions, gDNA isolation was performed using the Qiagen gDNA isolation kit (Cat. No. 13323). The sgRNA was amplified using the Emerald Amp Master Mix (Takara Code No. RR310A). Purified fragments were denatured and re-annealed by heating to 96°C for 5 minutes and then gradually cooled down to 4°C. Following the denaturation and re-annealing step, a population of renatured products was obtained, comprising one healthy strand (no mutation) and one strand with mutations, thereby creating a mismatch bulge. The DNA was then cleaved at the mismatch site, validating the deletion or addition of bases caused by the CRISPR-Cas9 system. Approximately 500 ng of re-annealed DNA was digested with 10 U of T7E1enzyme (Cat. No. M0302S) at 37°C for 20 minutes. The cleaved DNA product was visible on an agarose gel (Supplementary Fig. S4).
2.4. Vector Construction
For the engineering of cell lines, two plasmids, namely, Tagging and Donor plasmids, were constructed. These vectors were utilized for the generation of both acceptor and transgenic cell lines.
(i) Tagging Plasmid
The Tagging DNA was assembled using pTZ57R as a backbone, where we engineered various elements such as the promoter, reporter gene, resistance marker, poly (A) signal, and FRT sites from multiple vectors. The CMV enhancer, promoter, and EGFP were amplified from PeGFP, with the forward primer (FP) containing the FRT sequence and a KpnI restriction site, and the reverse primer (RP) containing the T2A sequence and a SalI restriction site. This construct was designated as Tagging clone 1. The Neo/Kan fragment and SV40 were amplified from pcDNA, where SalI in the FP and HindIII in the RP were used for cloning into the Tagging clone 1 plasmid. The RP was divided into two primers due to the length of the FRT sequence. The PCR product was digested with SalI and HindIII and ligated with the digested Tagging clone 1. Additionally, FRT and T2A sequences were introduced into the vector using primers. For cloning LHA and RHA into the tagging plasmid, multiple restriction sites (EcoRI, Eco53kI, SacI, Acc651, KpnI, AscI) were inserted into the FRT FP, and PacI, FseI, HindIII were inserted downstream to the FRT RP. Multiple cloning sites were provided for common restriction enzymes for insertion of DNA thereby, providing versatility for cutting and ligating, increasing the probability of successful gene insertion. This allowed for the insertion of AAVSI LHA and AAVS1 RHA between the EcoRI/KpnI and PacI/FseI restriction sites, respectively (Fig. 1A). The presence of GFP, Neomycin, LHA, and RHA was confirmed using restriction digestion and PCR (Supplementary Figs. S5 and S6). Finally, the clones were validated by Sanger sequencing and named as Tagging DNA.
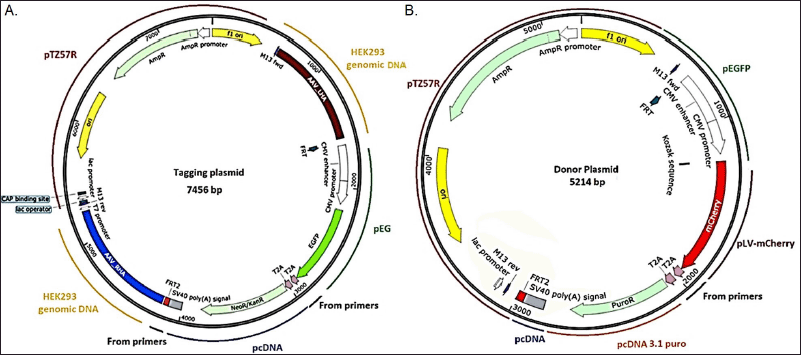 | Figure 1. Vector maps depicting the various regions of (A) Tagging plasmid and (B) donor plasmid. Parts of the plasmids are cloned from different vectors and genomic sequences. [Click here to view] |
(ii) Donor plasmid
The Donor Plasmid was constructed with the aim of generating a transgenic cell line or target cell line. It incorporates the mCherry gene, which was amplified from pLVmCherry using NheI RE in the FP and BamHI RE in the RP. The PCR product was subsequently cloned into the Tagging plasmid to construct the Donor plasmid. The clones were validated through Sanger sequencing, and the confirmed sequence was designated as the Donor plasmid.
An additional selection marker, Puromycin, was amplified from pcDNA 3.1puro and cloned into the Donor plasmid using SalI and BstBI enzymes. The clones were confirmed using restriction digestion (Supplementary Fig. S7) and Sanger sequencing. The positive clone was named the Donor plasmid (Fig. 1B).
2.5. Cell Lines and Reagents
HEK293, HeLa, MCF-7, Jurkat cells, HepG2, HT1080, and Caco2 cells were obtained from ATCC (Gaithersburg, MD, USA). Jurkat cell line was cultured in Roswell Park Memorial Institute (RPMI)-1640 medium supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin (P/S). HEK 293, MCF-7, HepG2, HT1080, and Caco2 were cultured in DMEM medium supplemented with 10% FBS and 1% P/S. DMEM, RPMI, FBS, Trypsin-EDTA, Puromycin, and P/S were from HiMedia.
2.6. Transfection of Cell Lines
To generate the acceptor and target cell lines, Hela, HEK293, HepG2, MCF-7, HT1080, and Caco2 cell lines were seeded into six-well plastic culture plates at a density of 2 × 105 cells/well in DMEM medium supplemented with 10% FBS. Concurrently, Jurkat cells were seeded in a T75 plastic flask at a density of 5 × 105 cells/well in RPMI medium supplemented with 10% FBS. This was done 1 day prior to transfection.
The transfection process was carried out using the BES buffer and CaCl2 method. 16–20 hours post-transfection, the medium was replaced, and the cells were allowed to proliferate in their respective media, supplemented with the appropriate antibiotic. This approach facilitated the growth and development of the cell lines under controlled conditions, ensuring their viability and effectiveness for subsequent procedures.
2.7. Acceptor Cell Line
To establish an acceptor cell line, a co-transfection was performed on the host cell lines (Hela, HEK293, HepG2, MCF-7, HT1080, CaCO2, and Jurkat) using the tagging plasmid and the gRNA-CRISPR-Cas9 expressing plasmid. To measure HR rates, 5 µg of tagging DNA plasmid and 5 µg of Cas9-expressing plasmid had been co-transfected to all the cell lines, followed by antibiotic selection. The experiment was then been repeated in triplicates (Supplementary Table 2). Following 48 hours post-transfection, the cells were subjected to selection using the neomycin antibiotic. The acceptor cell line was validated by assessing the expression of the reporter gene via flow cytometry, and cell sorting was conducted using fluorescence-activated cell sorting. Post cell sorting, the cells were allowed to proliferate in the presence of the appropriate antibiotic. A monoclonal cell line was obtained from the engineered cells using the limited dilution method (Supplementary Fig. S8). Upon obtaining a positive cell clone, the cell line was designated as the acceptor cell line (for instance, the HEK-293 cell line was named as the HEK293 acceptor cell line), selected, and routinely cultured in selective media (Fig. 2).
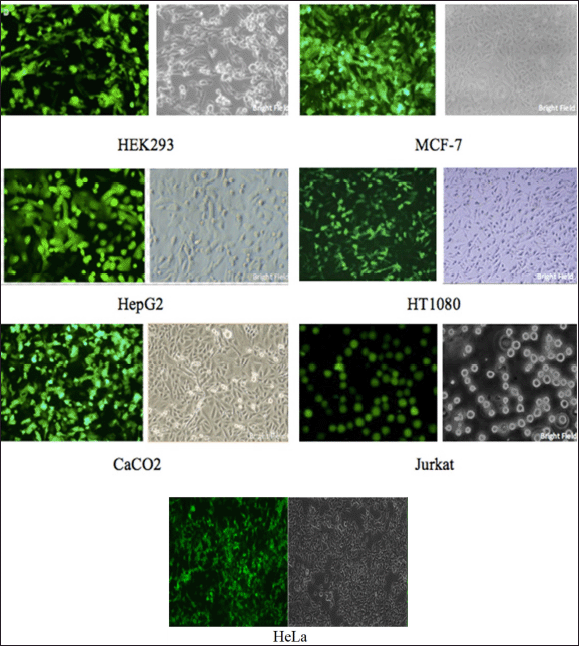 | Figure 2. Acceptor cell line confirmation. Representative images of the acceptor cell line mono clones post limited dilution method. The cells were selected using antibiotics to ensure avoiding non-transfected/ non-engineered cells. The images show uniform eGFP expression of all the cell lines. [Click here to view] |
2.8. Target Cell Line
All acceptor cell lines underwent co-transfection with 4 µg of the donor plasmid (10 µg) and 10 µg of pOG44 (Flp expressing plasmid), utilizing the BES buffer and CaCl2 method. This was followed by a selection process using puromycin (1 μg/ml) to generate a stable cell pool. Regular monoclonal cell lines were prepared using the same limited dilution method as the acceptor cell line. The experiment was performed in triplicates to ensure reliability. Surviving colonies had been assessed to determine efficiency (Supplementary Table 2). The positively selected stable cells were pelleted and seeded into a 12-well plate at a density of 50,000 cells in DMEM and RPMI-1640 medium, supplemented with 10% FBS per well. These cells were then incubated at 37°C with 5% CO2 overnight (16–24 hours). This process ensured the growth and development of the cell lines under controlled conditions, thereby facilitating their viability for subsequent procedures.
2.9. Clonal Isolation of Cell Lines
After transfection, clonal cell lines were isolated by serial dilution method followed by an expansion period using the University of Connecticut Storrs protocol [10]. Adherent cells underwent trypsinization, washing, and resuspension in their respective cell media. For the suspension cell line, cells were centrifuged and resuspended in an RPMI medium. Cells were seeded into a 96-well plate at a density of 4 × 104 cells/ml, with 100 µl of cells per well. These cells were then serially diluted by a 1:1 ratio to adjacent wells. An additional 100 µl of medium was added to each well, resulting in a total volume of 200 µl/well. The plates were incubated at 37°C in a humidified CO2 incubator. Colonies were detected under a microscope after a period of 7 to 12 days. These colonies were subsequently sub-cultured into 24 or 48-well plates for further growth (Supplementary Fig. S9).
2.10. Stable Cell Line Generation
The stability of various passages of acceptor cell lines and target cells was simultaneously monitored. All the target cell lines were expanded to produce sufficient freeze-downs for stability assessment. This was evaluated by thawing a given vial and passaging it 28 times to ensure uniform expression of the foreign genes (mCherry) throughout the passages. This rigorous process ensured the generation of stable, reliable cell lines for further experimentation and analysis. The cells were visualized using a Canon inverted microscope and images were captured with a 10× objective and 10× eyepiece magnification to achieve a total magnification of 100×.
2.11. Flow Cytometry
Adherent cell lines underwent digestion using 0.25% trypsin-EDTA and were subsequently suspended in their respective 10% serum-containing medium. In contrast, suspension cells (Jurkat) were collected into a 15 ml conical centrifuge tube and centrifuged at 300–400 g at room temperature for 10 minutes. The cell lines were maintained at a concentration of 1 × 105 cells/ml, as counted using a hemocytometer.
Samples were collected in PBS (free of Ca2+ and Mg2+) containing 1 μg/ml of Propidium Iodide (PI) (Thermo Fisher, P3566), and a flow cytometer (Thermo scientific Bigfoot spectral cell sorter) was employed to detect the GFP fluorescent signal (Becton Dickinson, San Jose, CA). The PI stain was utilized to differentiate between live and dead cells. Three samples were used to calculate the mean GFP fluorescence from 104 living cells per sample. The photomultiplier tube, which allows for the study of fluorescence at 530 nm, was used to quantify GFP fluorescence. Clumped cells were gated out based on their forward scatter and side scatter profiles. The target cell lines were confirmed using the same protocol, with fluorescence at Ex/Em 587/610 nm.
3. RESULTS AND DISCUSSION
CRISPR-Cas9 has revolutionized genetic engineering across many species. Initially aimed at understanding bacterial defense mechanisms, it now facilitates selective gene editing. CRISPR employs a gRNAs to target DNA, where Cas9 induces degradation [11,12]. The combination of CRISPR and HR enables flexible cassette exchange, allowing precise manipulation of the GOI [13].
3.1. Construction of Tagging Plasmid and Donor Plasmid
Five gRNAs were cloned into the PX459 plasmid, screened via a mismatch cleavage detection assay, and subsequently confirmed through Sanger sequencing (Supplementary Table 1). The mismatch cleavage detection assay was conducted using either Surveyor nuclease or T7E1, both of which induce a double-stranded break at the site of mismatch between two distinct strands of annealed DNA. Following the Surveyor nuclease assay, gRNA cleavage was validated by agarose gel electrophoresis (Supplementary Fig. S4). Out of the five gRNAs, one (5′-ACACCCCCATTTCACAGGTGAGG-3′) was selected for the generation of acceptor cell lines.
The Tagging DNA Plasmid was assembled via a two-step cloning process. In the first step, a tagging plasmid was constructed incorporating the CMV enhancer, CMV promoter, EGFP, and FRT sequence, and was designated as “tagging 1”. In the second step, the Neo/Kan fragment and SV40 polyA were cloned into the “tagging 1” plasmid, resulting in the final “Tagging plasmid”. Similarly, donor plasmids were constructed with an antibiotic selection marker (Puromycin) and mCherry as the GOI and were named “Donor puromycin plasmid” (Fig. 1).
3.2. Establishment of Acceptor Cell Lines and Target Cell Lines
The precision required for HR between exogenous and chromosomal DNA to generate healthy, viable cell lines is achieved through various approaches. These include the use of chimeric DNA-RNA oligonucleotides, triplex-forming oligonucleotides, and small single- or double-stranded oligonucleotides in conjunction with nonviral gene delivery methods for gene repair [14]. However, these techniques are limited to the correction of point mutations or small deletions/insertions. AAV vectors have been utilized as viral vectors for gene repair [15,16]in which cellular senescence and low conventional gene targeting rates limit experimental and therapeutic options. We have shown previously that linear, single-stranded DNA vectors based on adeno-associated virus (AAV, but their propensity for random integration may overshadow the benefits of targeted integration. Moreover, current AAV vectors are less efficient in transducing certain stem cell types, such as embryonic stem cells and hematopoietic cells, compared to adenoviral vectors [17,18]. Consequently, our protocol integrates the efficiency of CRISPR and AAV vectors against various cell lines.
Acceptor cell lines were generated by co-transfecting the tagging plasmid and the gRNA-CRISPR-Cas9 expressing plasmid into host cell lines (Hela, HEK293, HepG2, MCF-7, HT1080, CaCO2, Jurkat). Following 48 hours of transfection, cells were subjected to antibiotic (Neomycin) selection. The introduction of CRISPR, along with the AAVS1 homology sequence through a tagging plasmid, aided in the insertion of a gene at the AAVS1 site in the genome. The efficiency of this method ranged from 10% to 17% across all cell lines, thereby demonstrating the method’s uniformity across all cell lines (Supplementary Table 2). The acceptor cell line was validated by assessing the expression of the reporter gene via flow cytometry. A stable monoclonal cell line pool was obtained using a limited dilution method in a 96-well plate [10]. The cells were allowed to grow in the presence of antibiotics and were detected by microscopy after 8–10 days (Fig. 2). Cells were allowed to grow in the presence of antibiotics and were detected by microscopy after 8–10 days (Fig. 2). Upon obtaining a positive clone cell line, deemed as the “acceptor” cell line, they were routinely cultured in selective media. The positive stable cells were pelleted and seeded into a 12-well plate at a density of 50,000 cells in DMEM (for adherent cells) and RPMI-1640 medium (for suspension cells) with 10% FBS per well and incubated at 37°C, 5% CO2 overnight (16–24 hours).
Following the establishment of all acceptor cell lines (Hela, HEK293, HepG2, MCF-7, HT1080, CaCO2, Jurkat), they were co-transfected with the donor plasmid and pOG44 using the BES buffer and CaCl2 method and grown in the presence of puromycin (1 μg/ml) to generate a stable cell pool. The selected positive cells were pelleted and re-seeded into a 12-well plate at a density of 5 × 104 cells in DMEM and RPMI-1640 medium with 10% FBS per well and incubated at 37°C with 5% CO2 overnight (16–24 hours).
The acceptor cell line was successfully established using the tagging plasmid and validated CRISPRs that cleaved the AAVS1 locus. Consequently, the FRT tags are now inserted at the AAVS1 locus, thereby creating a provision to flip-in the GOI at the same locus. The mCherry sequence is strategically located between the FRT sites in the donor plasmid. The presence of mCherry was validated using PCR of the isolated gDNA in the target cell lines. Following transfection of the donor plasmid into the acceptor cell line, we observed mCherry fluorescence, indicating successful HR at the FRT sites. This result confirms that the integration of mCherry was not random but occurred at the intended genomic location. The cell line was selected by dual selection markers: GFP fluorescence and Neomycin, respectively. The purity of the cell line was assessed by flow cytometry, which showed ~100% GFP fluorescence (Fig. 3). Furthermore, PCR was used to confirm the presence of GFP, Neomycin marker, LHA, and RHA (Fig. 4).
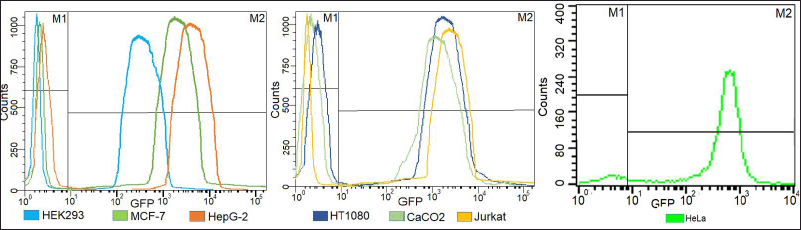 | Figure 3. Overlay flowcytometry plots of selected mono clones of acceptor cell lines showing >98% expression of eGFP as compared to control cell lines. [Click here to view] |
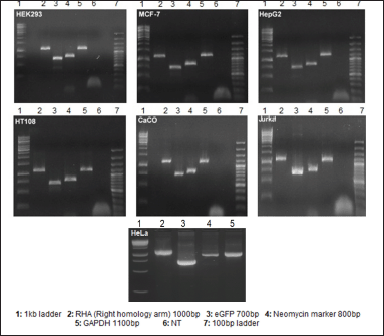 | Figure 4. PCR confirmation of the presence of tagging plasmid in the acceptor cell lines after total DNA isolation from transfected cell lines. It helps in confirming the integration of RHA and eGFP in the cell lines. GAPDH was used as a positive control. NT = No template. [Click here to view] |
3.3. Target Cell Conformation
Puromycin and mCherry, which served as the GOI, were employed as markers to validate the Target cell line as a proof of concept. Following the transfection of the donor plasmid, the target cell line underwent clonal selection via puromycin (1,000 μg/ml). Single-cell clones were then scaled up for the assessment of mCherry expression, utilizing flow cytometry (Fig. 5) and fluorescence microscopy (Fig. 6). The presence of both puromycin and mCherry confirmed the successful transfer of the complete gene fragment from the donor plasmid to the target cell line. Moreover, the absence of neomycin and eGFP, resulting from the “flipping out” of the tagging fragment from the acceptor cell line to create the target cell line, further validated the generation of the target cell line.
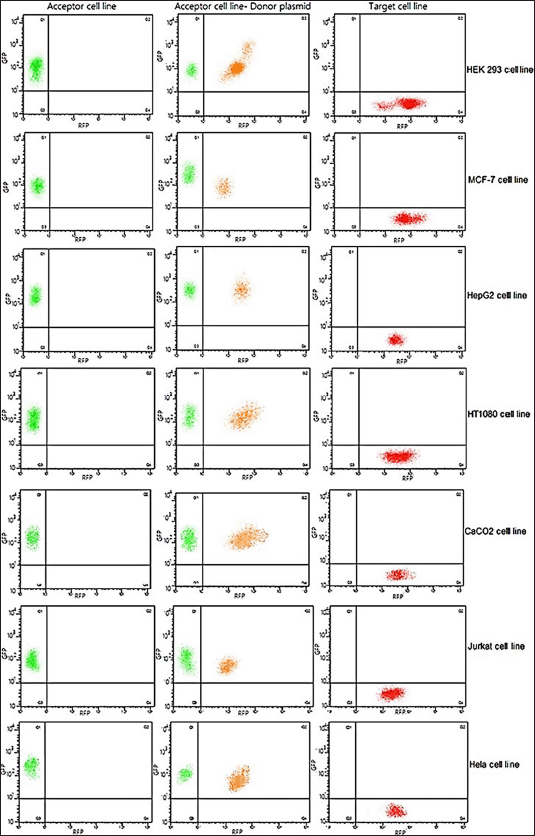 | Figure 5. Generation of target cell line. Representative flow-cytometry plots of acceptor, intermediate, and target cell lines. Left panel shows eGFP expression alone before transfection of the target gene using the donor plasmid. Middle panel shows a dual expression of mCherry and eGFP as an intermediate step. Right panel shows an exclusive expression of mCherry post 2nd antibiotic selection wherein transfected cells perish due to lack of newly introduced antibiotic marker (donor plasmid). [Click here to view] |
This recombination-mediated cassette exchange (RMCE) method encompassed Flp for recombining genes of interest such as mCherry gene in this case. This method shows an efficiency from 51% to 65%, demonstrating a huge rise in efficiency where half the cell number shows the insertion of a GOI in the host cell line (Supplementary Table 1). Concurrent evaluations for stability were conducted on the target cells and acceptor cell lines at various stages. Up to 28 passages were performed for the target cell lines, and their stability was confirmed through PCR amplification of mCherry and a housekeeping gene (GAPDH). The resulting amplicons indicated that the cell lines maintained relative stability (Supplementary Fig. S10).
3.4. Flow Cytometry
A complete flow cytometry profile of acceptor, intermediate, and target cell lines highlighting the cell numbers before tagging plasmid transfection (eGFP expression) is portrayed in Figure 5. The transition stage features dual mCherry and eGFP expression. After antibiotic selection, only mCherry is expressed as transfected cells without the antibiotic marker perish.
Our methodology integrates two genome engineering techniques: CRISPR-mediated HDR and FLP-FRT site-directed recombination technology. In this approach, a promoter and Frt site are introduced to a targeted region via a DSB induced by Cas9. Subsequently, a GOI is inserted into a predetermined sequence of a genomic site through Frt recombination. Upon correct insertion of an antibiotic resistance gene, it will express from a promoter and be integrated into the genome during the initial phase, thereby ensuring the selection of appropriate integrants. The selection cassette is then excised by a Flp-FRT recombination event, leaving behind a precisely targeted GOI. The Flp-FRT system, a widely employed site-directed recombination technology, originates from Saccharomyces cerevisiae. The Flp recombinase catalyzes DNA rearrangements using two FRT sites (Fig. 7).
 | Figure 6. Target cell line confirmation. Representative images of the target cell line mono clones post limited dilution method. Puromycin was used to select the cells, ensuring the exclusion of cells that were not transfected or engineered. The images demonstrate a consistent expression of mCherry. [Click here to view] |
 | Figure 7. Depiction of overall strategy for generating transgenic cell lines where M-cherry GOI is engineered into the host genome as an example. [Click here to view] |
3.5. Method Efficiency
In our procedure, we utilized CRISPR technology to strategically introduce FRT sites into a neutral locus within the genome via HR. This locus, being transcriptionally active yet non-coding, served as an ideal site for subsequent genetic manipulations. Following the establishment of these FRT sites, we could insert the GOI into the same locus through RMCE, by employing Flp.
Our method leverages the combined power of CRISPR and the AAVS1 homology sequence, facilitated by a tagging plasmid, to precisely target the insertion of the desired gene into the genome. Remarkably, this technique exhibits a consistent efficiency ranging from 10% to 17% across diverse cell lines (Fig. 8).
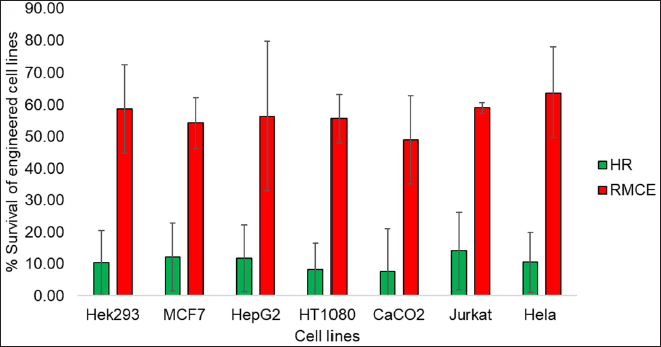
 | Figure 8. Chart depicting the overall survival of cell lines at HR mediated acceptor cell line formation and RMCE mediated transgenic cell line formation. [Click here to view] |
Central to our approach is the utilization of Flp for RMCE, enabling the efficient recombination of genes of interest, exemplified by the successful integration of the mCherry gene in our experiments. This aspect of our method yields impressive efficiencies ranging from 51% to 65%, representing a substantial improvement over traditional methods and highlighting its potential for widespread adoption (Fig. 8).
While HR may exhibit lower efficiency due to its rarity, RMCE offers a superior alternative, capitalizing on the FRT/FLP system to drive efficient recombination events. Once an “acceptor cell line” is established, the versatility of our approach allows for the insertion of multiple genes of interest through a streamlined single transfection process, bypassing the need for cumbersome two-plasmid transfections. These “packaged” cell lines can be used for generating engineered cell lines with ~65% efficiency in any laboratory harboring BSL2 facilities with a simple transfection protocol. We report a pan-cell line method for obtaining engineered cells with high efficiency.
Moreover, this method aligns with several United Nations Sustainable Development Goals (SDGs). By advancing medical research and biotechnology, our work contributes to SDG 3 (good health and well-being), with the potential to develop new treatments and therapies for various diseases. The precision genetic manipulation techniques we have developed foster innovation in biotechnology and genetic engineering, thereby supporting SDG 9 (industry, innovation, and infrastructure). Furthermore, the research promotes higher education and research opportunities in the fields of genetics, biotechnology, and medical sciences, aligning with SDG 4 (quality education). Additionally, our approach to creating transgenic cell lines with precision can lead to more efficient and sustainable practices in genetic research and biotechnology, contributing to SDG 12 (responsible consumption and production). By framing our research within the context of these SDGs, we demonstrate its broader impact and relevance to global sustainability efforts.
4. CONCLUSION
We introduce a highly efficient approach for the generation of target cell lines, leveraging the combined power of CRISPR-mediated HDR and FLP-FRT site-directed recombination technology. The effectiveness of this method has been established across a variety of cell lines, including HeLa, HEK293, MCF-7, Jurkat, HepG2, HT1080, and Caco2, representing a diverse array of human tissues and underscoring the robustness of the developed platform.
The concurrent introduction of CRISPR, alongside the AAVS1 homology sequence facilitated by a tagging plasmid, serves to precisely guide the insertion of the GOI into the designated AAVS1 site within the genome. Notably, the efficacy of this methodology manifests consistently, exhibiting a notable efficiency ranging from 10% to 17% across a spectrum of cell lines, thereby attesting to its uniform applicability. Central to the RMCE procedure is the strategic employment of Flp, which enables the judicious recombination of genes of interest, exemplified herein by the successful integration of the mCherry gene. Remarkably, this refined methodology yields an efficiency ranging from 51% to 65%, marking a substantial augmentation in efficacy, wherein nearly half of the cellular population evidences the insertion of the GOI within the host cellular milieu.
The versatility and precision of this method demonstrated through the selective insertion and excision of genes of interest, highlight its potential for widespread applications in genetic engineering and related fields. The findings of this study significantly contribute to the advancement of genome engineering methods, paving the way for more precise and efficient genetic manipulation.
5. AUTHOR CONTRIBUTIONS
All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; agreed to submit to the current journal; gave final approval of the version to be published; and agree to be accountable for all aspects of the work. All the authors are eligible to be an author as per the international committee of medical journal editors (ICMJE) requirements/guidelines.
6. FUNDING
There is no funding to report.
7. CONFLICTS OF INTEREST
The authors report no financial or any other conflicts of interest in this work.
8. ETHICAL APPROVALS
This study does not involve experiments on animals or human subjects.
9. DATA AVAILABILITY
All the data is available with the authors and shall be provided upon request.
10. PUBLISHER’S NOTE
All claims expressed in this article are solely those of the authors and do not necessarily represent those of the publisher, the editors and the reviewers. This journal remains neutral with regard to jurisdictional claims in published institutional affiliation.
11. USE OF ARTIFICIAL INTELLIGENCE (AI)-ASSISTED TECHNOLOGY
The authors declares that they have not used artificial intelligence (AI)-tools for writing and editing of the manuscript, and no images were manipulated using AI.
12. SUPPLEMENTARY MATERIAL:
The supplementary material can be accessed at the journal's website: Link here [https://jabonline.in/admin/php/uploadss/1285_pdf.pdf].
REFERENCES
1. Urnov FD, Rebar EJ, Holmes MC, Zhang HS, Gregory PD. Genome editing with engineered zinc finger nucleases. Nat Rev Genet 2010;11(9):636–46. CrossRef
2. Joung JK, Sander JD. TALENs: a widely applicable technology for targeted genome editing. Nat Rev Mol Cell Biol 2012;14(1):49–55. CrossRef
3. Hsu PD, Lander ES, Zhang F. Development and applications of CRISPR-Cas9 for genome engineering. Cell 2014;157:1262–78. CrossRef
4. Sakuma T, Woltjen K. Nuclease-mediated genome editing: at the front-line of functional genomics technology. Dev Growth Differ 2014;56:2–13. CrossRef
5. Aslam B, Rasool M, Idris A, Muzammil S, Alvi RF, Khurshid M, et al. CRISPR-Cas system: a potential alternative tool to cope antibiotic resistance. Antimicrob Resist Infect Control 2020;9:1–3. CrossRef
6. Chang HHY, Pannunzio NR, Adachi N, Lieber MR. Non-homologous DNA end joining and alternative pathways to double-strand break repair. Nat Rev Mol Cell Biol 2017;18(8):495–506. CrossRef
7. Heigwer F, Kerr G, Boutros M. E-CRISP: fast CRISPR target site identification. Nat Methods 2014;11(2):122–3. CrossRef
8. Tiyaboonchai A, Mac H, Shamsedeen R, Mills JA, Kishore S, French DL, et al. Utilization of the AAVS1 safe harbor locus for hematopoietic specific transgene expression and gene knockdown in human ES cells. Stem Cell Res 2014;12:630. CrossRef
9. Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. Genome engineering using the CRISPR-Cas9 system. Nat Protoc 2013;8(1):2281–308. CrossRef
10. Freshney RI. Culture of animal cells: a manual of basic technique and specialized applications. 6th edition, John Wiley & Sons Ltd., Hoboken, NJ, 2011 CrossRef
11. Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012;337:816–21. CrossRef
12. Tran NT, Bashir S, Li X, Rossius J, Chu VT, Rajewsky K, et al. Enhancement of precise gene editing by the association of Cas9 with homologous recombination factors. Front Genet 2019;10:365. CrossRef
13. Zhang C, Xiao B, Jiang Y, Zhao Y, Li Z, Gao H, et al. Efficient editing of malaria parasite genome using the CRISPR/Cas9 system. mBio 2014;5(4):e01414–14. CrossRef
14. Al-Dosari MS, Gao X. Nonviral gene delivery: principle, limitations, and recent progress. AAPS J 2019;11:671–81. CrossRef
15. Chamberlain JR, Schwarze U, Wang PR, Hirata RK, Hankenson KD, Pace JM, et al. Gene targeting in stem cells from individuals with osteogenesis imperfecta. Science 2004;303:1198–201. CrossRef
16. Hirata R, Chamberlain J, Dong R, Russell DW. Targeted transgene insertion into human chromosomes by adeno-associated virus vectors. Nat Biotechnol 2002;207(20):735–8. CrossRef
17. Smith-Arica JR, Thomson AJ, Ansell R, Chiorini J, Davidson B, McWhir J. Infection efficiency of human and mouse embryonic stem cells using adenoviral and adeno-associated viral vectors. Cloning Stem Cells 2003;5(1):51–62. CrossRef
18. Zhong L, Li W, Yang Z, Qing K, Tan M, Hansen J, et al. Impaired nuclear transport and uncoating limit recombinant adeno-associated virus 2 vector-mediated transduction of primary murine hematopoietic cells. Hum Gene Ther 2004;1(12):1207–18. CrossRef