
1. INTRODUCTION
Colorectal cancer (CRC) is a malignant tumor of the digestive tract that arises from the abnormal proliferation of intestinal epithelial cells, leading to sustained malignant transformation [1]. It has become the third most common cancer in the world, with a mortality rate ranking third in males and second in females [2]. The majority of colorectal tumors are initially caused by familial adenomatous polyposis (FAP) [3], an autosomal dominant hereditary disease resulting from mutations in the adenomatous polyposis coli gene [4], with an incidence of approximately 1/7,000–1/30,000 [5]. If left untreated, almost 100% of FAP patients will develop CRC[6].
Apc gene-encoded protein is a tumor suppressor that promotes the maintenance of degradation homeostasis of β-catenin, a critical effecter molecule in the Wnt signaling pathway [7], through negative regulation by binding to it. Loss or reduction of Apc gene function leads to the accumulation of free β-catenin in the cytoplasm and its translocation to the nucleus, activating the Wnt pathway and related oncogenes such as KRAS mutations or inactivated tumor suppressor gene TP53 [8,9]. Common forms of Apc gene mutations include frameshift, nonsense, splice site abnormality, missense, and large segment deletion, leading to the production of non-functional proteins [10,11]. Multiple CRCs are characteristic of FAP, where Apc gene truncation or mutation is inherited, while another allele may be lost in adenomas [5,12]. Homozygous deletion of the Apc gene is very rare [13], and Apc gene abnormalities are the main cause of FAP and malignant tumor development [14,15].
The pathogenesis of CRC is complex, and changes in the composition of gut microbiota are one of the contributing factors. The intestinal microbiota, which is composed of a large number of microorganisms [16], is present in the colon. It was suggested that dysbiosis of the gut microbiota is closely related to CRC, and the role of microbiota in initiating and promoting CRC is becoming increasingly apparent [16]. Recent studies have identified several bacteria as candidate pathogens for CRC, including Streptococcus bovis, Bacteroides fragilis, Fusobacterium nucleatum, Escherichia coli, and anaerobic bacteria. The mechanisms by which gut microbiota is associated with CRC mainly include inflammatory response, immune regulation, dietary components, metabolism, and genotoxins [17,18].
In 1990, the ApcMin/+ mouse model was first established by Dove Lab of Wisconsin-Madison University. The researchers induced C57BL/6J male mice using the mutagen ethylnitrosourea, followed by crossbreeding with AKR/J female mice. They observed that a subset of the offspring was prone to developing intestinal tumors, in which the homologous Apc gene at amino acid position 850 was mutated from Leu to a stop codon, leading to premature termination of translation and the development of multiple intestinal adenomas [19]. In 1997, the first conditional Apc functional deficiency model was developed [20], leading to the occurrence of adenomas in the colon. To construct more models for late-stage diseases, the ApcMin/+ mouse model was developed and involved in investigating the common mutations of oncogenes or tumor suppressor genes and their related genes [21].
In this study, we employed the highly efficient CRISPR/Cas9 gene editing technique, to introduce a mutation targeting the Apc-202 transcript (ENSMUST00000079362.12) in vivo within the mouse model through homology-directed repair. Specifically, we introduced the mutation target site to replace the amino acid Leu at the 850th position of the mouse Apc gene with a stop codon TAG (TTG to TAG). As a result, we successfully established the ApcL850X point mutation mouse model and conducted preliminary phenotype validation of spontaneous intestinal cancer. We investigated the alterations in the gut microbiota of mice with spontaneous CRC to explore potential correlations between CRC and gut microbiota, thereby contributing to the development of microbiome-targeted therapies and CRC prevention strategies in the future.
2. MATERIALS AND METHODS
2.1. Mice
In all experiments, mice were maintained in a specific pathogen-free environment, with ad libitum access to food and water under a 12-hour light/dark cycle. All procedures employed in this study were approved by the Institutional Animal Care and Use Committee of Shanghai Laboratory Animal Research Center (Authorization numbers: 2211028081) and were conducted in accordance with relevant national and international guidelines.
2.2. Generation of C57BL/6-ApcL850X Point Mutation Mice
C57BL/6-ApcL850X point mutation mice, abbreviated as ApcL850X mice below, were generated by Shanghai Model Organisms Center, Inc. (SMOC, Shanghai, China). This model was developed by CRISPR/Cas9-mediated gene editing in zygotes.
The mouse Apc gene is located on chromosome 18 (Chromosome 18: 34,354,037–34,455,605) and the Apc-202 transcript is approximately 12.811 kb in length, consisting of 16 exons and 15 introns, with a total of 2,842 amino acids. Based on the structure of the mouse Apc gene, the mutation site and gRNA target were selected in the coding region of exon 16. The gRNA target sequence was 5′-TTCTGAGAAAGACAGAAGTT TGG-3′. An oligo donor DNA was synthesized for the construction of the gRNA expression vector, with a sequence of 5′-GACTGTTCTTTCACCATATTTAAATACTACGGTATTGCCCAGCTCTTCTTCCTCAAGGGGAAGTTTAGACAGTTCTCGTTCTGAGAAAGACAGAAGTTAGTAGAGAGAGAGCGAGGTATTGGCCTCAGTGCTTACCATCCAACAACAGAAAATGCAGGAACCTCATCAAAACGAGGTCTGCAGATCACTACCACTGCAGCCCAGATAGCCAA-3′. The gRNA expression vector pX-T7-sgRNA was linearized by enzyme digestion and transcribed in vitro according to the instructions of the HiScribe T7 High Yield RNA Synthesis Kit (New England Biolabs, MA). The Cas9 expression vector was linearized by XbaI digestion and transcribed in vitro according to the instructions of the mMESSAGE mMACHINE T7 Ultra Transcription Kit (Thermo Fisher Scientific, MA).
Cas9 mRNA, gRNA, and oligo donor DNA were mixed and microinjected into the fertilized eggs of C57BL/6 wild-type mice (WT mice). Subsequently, the injected embryos were transferred into the uteri of pseudo-pregnant Institute of Cancer Research mice female mice to obtain F0 generation mice. F1 generation mice, which were heterozygous for the Apc gene point mutation (ApcL850X), were generated by crossing the F0 positive mice with WT mice. Genotyping of F1 mice was performed using the primers targeting the P1 (sequences: 5′-CATCCCTTCACGTTAGGAAACAG-3′) and P2 (sequences: 5′-CTTTGGCATAAGGCATAGAGCAT-3′). The PCR conditions consisted of an initial denaturation at 94°C for 3 minutes, followed by 35 cycles of denaturation at 98°C for 15 seconds, annealing at 58°C for 15 seconds, extension at 68°C for 1 minutes, and a final extension at 68°C for 5 minutes. The PCR products were subjected to T-vector cloning and subsequently sequenced using the sequencing primer sequences performed with P1 (sequences: 5′-CATCCCTTCACGTTAGGAAACAG-3′). F2 generation ApcL850X mice heterozygous for the Apc gene point mutation were obtained by crossing F1 heterozygous mice with WT mice. The genotyping strategy for F2 mice was the same as for F1 mice.
2.3. Measurement of Body Weight, Food Intake, and Survival Curve
From postnatal day 20 to 150, daily observations were made to record the time of death for each mouse, while weight and food intake were measured and recorded weekly starting from 10 weeks of age. A total of 40 mice were used in this study, with 16 males and 16 females (n = 16 per sex). At the beginning of each week, a pre-weighed amount of standard laboratory chow was provided to each mouse in its individual cage, and the initial weight was recorded. At the end of the week, the remaining food, including scattered pieces, was collected and weighed. The weekly food intake for each mouse was calculated by subtracting the final weight of the remaining food from the initial weight provided. The weekly food intake data, along with body weight measurements, were systematically recorded for each mouse. These records were used to analyze trends in food consumption and correlate them with body weight changes and survival rates.
2.4. Routine Blood Test
Blood samples were collected from the cheek vein plexus of 18-week-old ApcL850X and WT mice under isoflurane gas anesthesia for routine blood examination. A total of 12 mice were used for this analysis, with 6 ApcL850X mice and 6 WT mice (n = 6 per group). The differences between ApcL850X mice and WT mice were analyzed and compared based on various blood parameters.
2.5. Tumor Count and Histopathological Analysis
The number of intestinal tumors and histopathological changes were assessed in mice euthanized by cervical dislocation at 120 days of age. A total of 40 mice were used for this analysis, with 13 males and 13 females (n = 13 per sex). The small intestine and colon were dissected, and the tumor count was recorded. Paraffin-embedded samples were processed for hematoxylin and eosin staining for histopathological observation.
2.6. Gut Microbiota Composition Preparation
Feces samples were collected once a week from 3 to 25-week-old mice, comprising a total of 10 ApcL850X female mice, 10 ApcL850X male mice, 10 WT female mice, and 10 WT male mice (n = 10 per group). The samples were immediately stored at −80°C until further analysis. Microbial community genomic DNA was extracted from feces samples using the E.Z.N.A.® soil DNA Kit (Omega Bio-tek, GA, US) according to the manufacturer’s instructions. The hypervariable region V3-V4 of the bacterial 16S rRNA gene was amplified with primer pairs 338F (5′-ACTCCTACGGGAGGCAGCAG-3′) and 806R(5′-GGACTACHVGGGTWTCTAAT-3′) by an ABI GeneAmp® 9700 PCR thermocycler (ABI, CA, USA). Purified amplicons were pooled in equimolar and paired-end sequenced on an Illumina MiSeq PE300 platform/NovaSeq PE250 platform (Illumina, San Diego, USA) according to the standard protocols by Majorbio Bio-Pharm Technology Co. Ltd. (Shanghai, China). The raw reads were deposited into the NCBI Sequence Read Archive database.
2.7. Processing of Sequencing Data
The raw 16S rRNA gene sequencing reads were demultiplexed, quality-filtered by fastp version 0.20.0 [22], and merged by FLASH version 1.2.7 [23] with the following criteria: (i) the 300 bp reads were truncated at any site receiving an average quality score of <20 over a 50 bp sliding window, and the truncated reads shorter than 50 bp were discarded, reads containing ambiguous characters were also discarded; (ii) only overlapping sequences longer than 10 bp were assembled according to their overlapped sequence. The maximum mismatch ratio of the overlap region was 0.2. Reads that could not be assembled were discarded; (iii) samples were distinguished according to the barcode and primers, and the sequence direction was adjusted, exact barcode matching, 2 nucleotide mismatch in primer matching.
Operational taxonomic units (OTUs) with a 97% similarity cut-off [24,25] were clustered using UPARSE version 7.1 [24], and chimeric sequences were identified and removed. The taxonomy of each OTU representative sequence was analyzed by RDP Classifier version 2.2 [26] against the 16S rRNA database (e.g., Silva v138) using a confidence threshold of 0.7.
2.8. Combined receiver operating characteristic (ROC) Analysis
To evaluate the diagnostic accuracy of gut microbiota composition in predicting CRC development, a combined ROC analysis was performed using data from 25-week-old female ApcL850X mice and 23-week-old male ApcL850X mice, compared with age-matched WT mice (n = 10 per group). The aim was to identify differences in microbiota composition between these groups at their respective ages.
Fecal samples were collected from 10 ApcL850X female mice, 10 ApcL850X male mice, 10 WT female mice, and 10 WT male mice. These samples were used to analyze the gut microbiota composition through high-throughput sequencing. The sequencing data were normalized using relative abundance to account for differences in sequencing depth. Data from the selected species were averaged to provide a comprehensive overview of microbiota composition. ROC curve analysis was performed to determine the sensitivity true positive rate (TPR) and specificity false positive rate (FPR) of different bacterial taxa as potential biomarkers for CRC. The area under the ROC curve (AUC) was calculated to assess the overall performance of the diagnostic model. The confidence interval (CI) threshold was set at 0.95.
Based on the phenotypic ApcL850X mice and WT mice, bacterial abundance data from high-throughput sequencing results were used to create a binary classification label (CRC vs. non-CRC). We used R software (version 3.3.1) with the plotROC package. This package allows for the generation of ROC curves and the calculation of AUC values, facilitating the comparison of different biomarkers' diagnostic performance.
2.9. Statistical Analysis
Statistical analysis was performed using GraphPad 8.0 software. All data were presented as Mean ± SEM. Group comparisons were analyzed using the t-test, and statistical significance was considered when p < 0.05.
3. RESULTS
3.1. Generation of C57BL/6-ApcL850X Point Mutation Mice
The construction strategy for ApcL850X mice is shown in Figure 1a. Cas9 mRNA and gRNA were obtained through in vitro transcription, and oligo donor DNA was synthesized. The microinjected embryos were transferred to pseudo-pregnant mice, and the newborn mice were designated as F0 generation. The genotyping strategy for F0 generation is shown in Figure 1a. Positive F0 mice were bred with WT C57BL/6 mice to obtain F1 generation point mutation heterozygous mice. Positive F1 mice were bred with WT C57BL/6 mice to obtain F2 generation point mutation heterozygous mice, while homozygous positive mice were lethal, and PCR analysis followed by sequencing confirmed the presence of the heterozygous mutant (ApcL850X mice). The specific sequence information after the point mutation is depicted in Figure 1b. The electrophoretic map for genotype identification results is presented in Figure 1c. The representative sequencing diagram of PCR reaction product results is shown in Figure 1d.
 | Figure 1. Construction of C57BL/6-ApcL850X point mutation mice. (a) Point mutation strategy, Cas9/gRNA, and oligo donor DNA complexes point mutate the Apc gene by recognizing a target in the exon16 region; (b) Point mutation site in ApcL850X mice; (c) is the electrophoretic map for genotype identification. The band size of WT mice was 612 bp, and that of heterozygous mice was 614 bp; (d) Representative sequencing of PCR reaction products, with a mutation stop codon in the red box. [Click here to view] |
3.2. Measurement of Body Weight, Food Intake, and Survival Curve
Starting at 10 weeks of age, the weekly changes in body weight were documented. Figure 2a and b illustrated that from 10 to 18 weeks of age, female ApcL850X mice exhibited a significant decrease in body weight compared to their WT counterparts of the same sex, with the disparity becoming apparent at 11 weeks (*p < 0.05, ** p < 0.01). In contrast, male ApcL850X mice showed lower body weights starting at 14 weeks. The study commenced recording weekly variations in food consumption at 11 weeks of age, revealing discernible distinctions between male ApcL850X mice and male WT mice beginning at 16 weeks. Conversely, no statistically significant variances were noted between female ApcL850X mice and the same-sex WT mice. The survival analysis indicated a stark contrast in outcomes, with all male ApcL850X mice succumbing by 23 weeks, while only 15% of female ApcL850X mice survived.
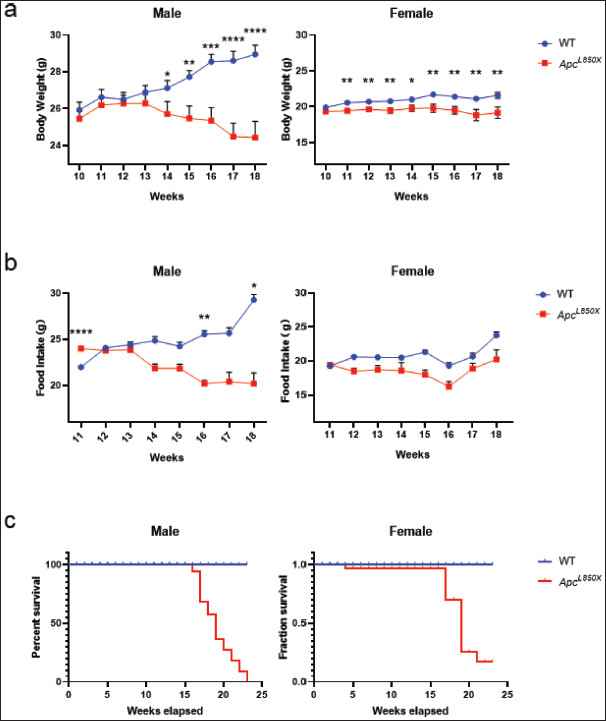 | Figure 2. Body weight, food intake, and survival curves of ApcL850X mice. (a) Changes in body weight of ApcL850X mice and WT mice from 10 to 18 weeks of age; (b) Food intake of ApcL850X mice and WT mice from 11 to 18 weeks of age; (c) Survival curves of ApcL850X mice and WT mice. * p < 0.05, ** p < 0.01,*** p < 0.001, n = 16. [Click here to view] |
3.3. Routine Blood Test
As shown in Figure 3, compared to WT mice, ApcL850X mice exhibited significant increases in WBC, MCV, PDW, MPV, NEUT#, MONO# (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001). On the other hand, ApcL850X mice showed significant decreases in RBC, HGB, MCHC, LYMPH%, and EO% (**p < 0.01, **** p < 0.0001). No relevant indicators related to reticulocytes were detected in ApcL850X mice. The results indicated that ApcL850X mice were experiencing symptoms of anemia and concurrently, there was inflammation present in their bodies.
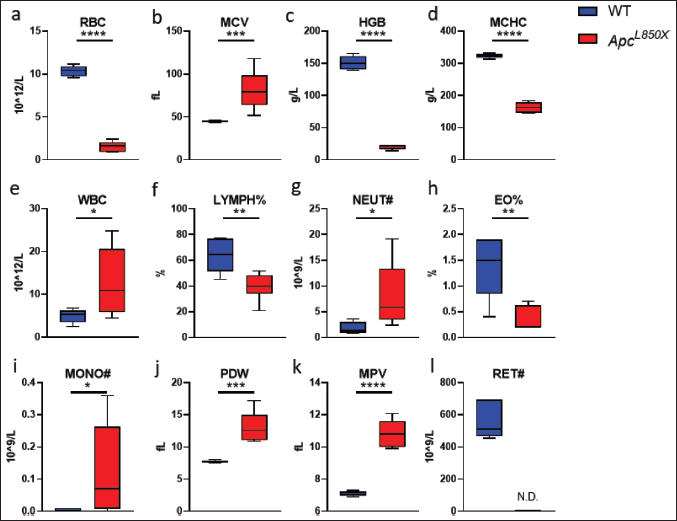 | Figure 3. Blood routine test of ApcL850X mice. (a) Red blood cell count (RBC): 10^12/l; (b) Mean corpuscular volume (MCV): fl ; (c) Hemoglobin (HGB): g/l; (d) Mean corpuscular hemoglobin concentration (MCHC): g/l; (e) White blood cell count (WBC): 10^9/l; (f) Lymphocyte percentage (LYMPH%): g/l ; (g) Neutrophil count (NEUT#): 10^9/l ; (h) Eosinophils (EO%); (i) Mononucleosis (MONO#); (j) Platelet distribution width (PDW); (k) Mean platelet volume (MPV); (l) Reticulocyte (RET#): 10^9/l. * p < 0.05, ** p < 0.01,*** p <0.001, n = 6. [Click here to view] |
3.4. Tumor Count and Histopathological Analysis
As depicted in Figure 4, the tumors within the intestinal tract of ApcL850X mice predominantly developed in the ileum, with the duodenum being the secondary site. Among these, the number of tumors in female mice slightly surpassed that in male mice; however, no statistically significant difference in tumor count was observed between male and female mice.
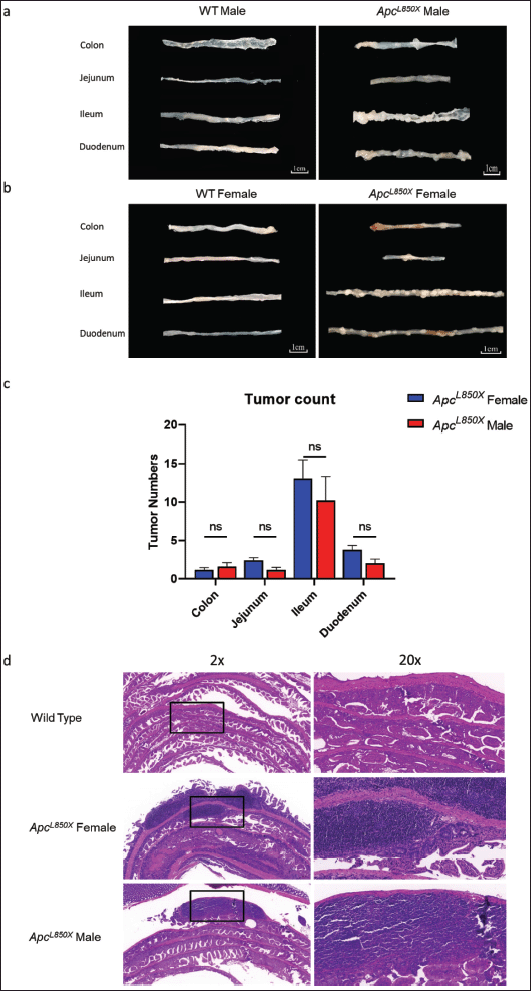 | Figure 4. The tumor count of intestinal tissues and pathology result. (a) intestinal samples from male ApcL850X mice and male WT mice; (b) intestinal samples from female ApcL850X mice and female WT mice; (c) The number of tumors in the colon, duodenum, jejunum, and ileum of female and male ApcL850X mice, n = 13; (d) HE staining of intestinal sections. [Click here to view] |
As shown in Figure 4d, no tumors were found in the intestinal Swiss rolls of WT mice. In contrast, the intestinal Swiss rolls of ApcL850X mice exhibited a higher number of polyps and areas of malignant transformation. The tumor infiltrated the muscular layer of the intestinal wall and invaded the surrounding tissues such as the serosa. The tumor cells exhibited abnormal morphology, increased nuclear division activity, cellular pleomorphism, and exhibited an abnormal tumor response.
3.5. Gut Microbiota Composition
As shown in Figure 5, there was a clear separation in the gut microbiota between ApcL850X and WT mice from 22 to 25 weeks of age. Figure 5a and b showed species diversity, which described the degree of diversity of microbial species in the gut microbiota. These figures indicated a significant difference in the diversity of gut microbiota between ApcL850X and WT mice.
 | Figure 5. Community diversity analysis. (a) Sample microbiota profiling analysis of ApcL850X mice and WT mice; (b) Sample level clustering tree of ApcL850X mice and WT mice; (c) The relative abundance of various microorganisms in the samples of ApcL850X mice and WT mice aged 22–25 weeks, n = 10. [Click here to view] |
Figure 6a The relative abundance of various microorganisms in the gut microbiota of female mice aged 22–25 weeks. Significant differences in the community structure and dominant species were observed between ApcL850X and WT mice. Specifically, after disease onset, ApcL850X female mice exhibited a notable absence of Lactobacilli and Dubosiella, along with an increase in Bacteroides, Alistipes, Akkermansia, and unclassified Oscillospiraceae.
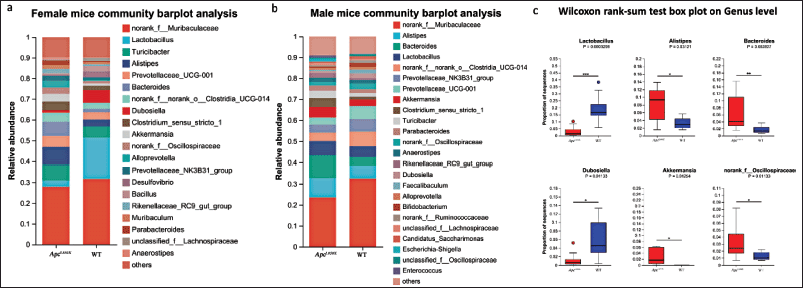 | Figure 6. Gut microbiota composition analysis. (a) The relative abundance of various microorganisms in the samples of female ApcL850X mice and female WT mice aged 22–25 weeks; (b) The relative abundance of various microorganisms in the samples of male ApcL850X mice and male WT mice aged 22–23 weeks; (c) Statistical comparison of the proportions of various bacteria in the gut microbiota of ApcL850X mice and WT mice aged 22–25 weeks. * p < 0 .05, ** p < 0.01, *** p < 0.001, n = 10. [Click here to view] |
Figure 6b The relative abundance of various microorganisms in the gut microbiota of male mice aged 22–25 weeks. Similar significant differences in community structure and dominant species were observed between ApcL850X and WT male mice.
The box plots in Figure 6c Statistical comparison of the proportions of various bacteria in the gut microbiota of ApcL850X and WT mice aged 22–25 weeks. The top six bacterial species showing statistically significant differences were Lactobacillus, Alistipes, Bacteroides, Dubosiella, Akkermansia, and unclassified Oscillospiraceae. The proportion of Lactobacillus and Dubosiella in ApcL850X mice feces was significantly lower than in WT mice, while the proportions of Alistipes, Bacteroides, Akkermansia, and unclassified Oscillospiraceae were significantly higher.
Figure 7a presented the alterations in dominant species, relative abundances, and community structure within the gut microbiota of ApcL850X mice aged 3–25 weeks. These data highlighted notable variations in the gut microbiota composition and dominant species with age in ApcL850X mice. Specifically, as the disease progresses, a decline in the proportions of Lactobacillus and Bifidobacterium was observed, accompanied by an increase in the abundance of Alistipes.
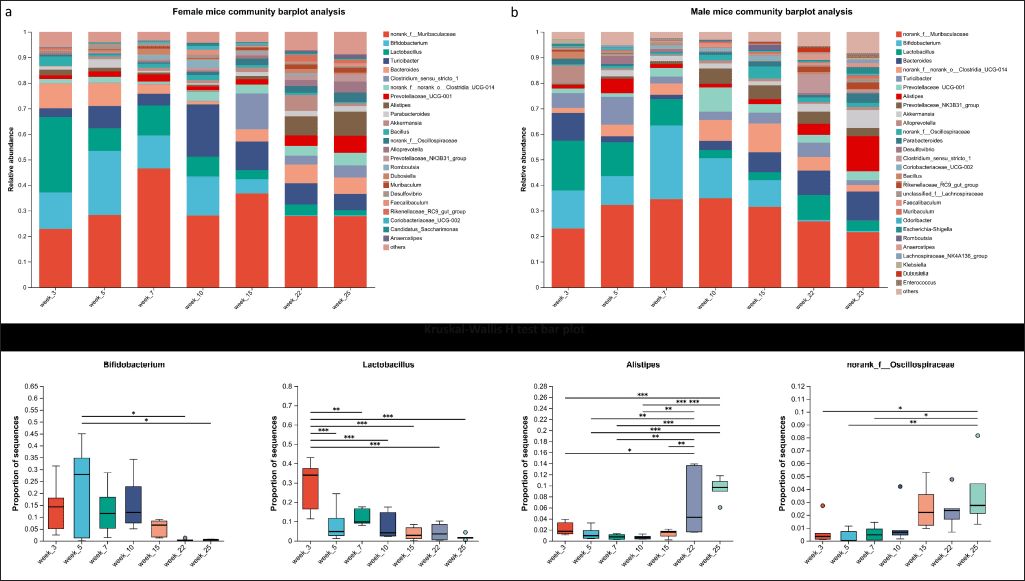 | Figure 7. Gut microbiota composition analysis changes. (a) The relative abundance of various microorganisms in the samples of female ApcL850X mice aged 3–25 weeks; (b) The relative abundance of various microorganisms in the samples of male ApcL850X mice aged 3–23 weeks; (c) Statistical comparison of the proportions of various bacteria in the gut microbiota of ApcL850X mice and WT mice aged 3–25 weeks, * p < 0.05, ** p < 0.01, *** p < 0.001, n = 5. [Click here to view] |
Figure 7b illustrated similar analyses for male mice. Significant differences in the gut microbiota composition and community structure were observed between male ApcL850X and WT mice at different ages. Notable shifts included a decrease in Lactobacillus and Bifidobacterium and an increase in Alistipes and norank_of__Oscillospiraceae.
The box plots at the bottom in Figure 7c showed the results of the Kruskal-Wallis H test, further highlighting the significant differences in the relative abundances of these bacterial taxa across different ages in both male and female mice.
3.6. COMBINED ROC ANALYSIS
To evaluate the diagnostic accuracy of gut microbiota composition in predicting CRC development, a combined ROC analysis was performed using data from 25-week-old female ApcL850X mice and WT mice, as well as 23-week-old male ApcL850X mice and WT mice. The aim was to identify differences in microbiota composition between these two groups at the same age.
The ROC curve for female mice in Figure 8a showed an AUC of 0.72 with a 95% CI of 0.48–0.96. The curve plots sensitivity (TPR) versus 1-specificity (FPR), with the points on the curve indicating the optimal threshold and the values in parentheses indicating the specificity and sensitivity corresponding to these points.
 | Figure 8. Combined ROC analysis of gut microbiota composition for CRC prediction. The ROC curves for (a) female mice and (b) male mice plot the sensitivity (TPR) against 1-specificity (FPR). The X-axis represents 1-specificity, ranging from 0 to 1, and the Y-axis represents sensitivity, also ranging from 0 to 1. Points marked on the curves indicate the optimal threshold values, with the values in parentheses representing the corresponding specificity and sensitivity at those points, n = 10. [Click here to view] |
The ROC curve for mice in Figure 8b showed an AUC of 0.67 with a 95% CI of 0.41–0.93, indicating a lower level of diagnostic accuracy compared to female mice. Similar to the ROC curve for females, this curve also plotted sensitivity versus 1-specificity, with optimal threshold values marked along the curve.
4. DISCUSSION
In our study, we employed the CRISPR/Cas9 system to introduce a point mutation in the Apc gene of mice by homologous recombination repair within exon 16, replacing the 850th amino acid residue (Leu) to a stop codon, resulting in an Apc gene missense mutation. This mutation caused the loss of tumor suppressor function. Furthermore, this mouse strain exhibited genetic stability during subsequent breeding and conservation processes, providing evidence that our design strategy was an efficient and rapid method for obtaining mice with point mutations in the Apc gene.
The phenotypic validation of ApcL850X mice revealed significant differences in body weight and food consumption between male ApcL850X mice and their male WT counterparts. The observed phenomenon can be attributed to the deteriorating health status of the ApcL850X mice post disease onset, leading to reduced appetite. The decrease in food intake subsequently resulted in a decline in body weight. In comparison to female WT mice, ApcL850X female mice exhibited a decrease in body weight without significant changes in food consumption. Furthermore, female ApcL850X mice showed a relatively milder trend of body weight reduction compared to male ApcL850X mice. Survival curve analysis showed that all male ApcL850X mice died at 23 weeks, while female ApcL850X mice survival was 15%. Based on the differences observed in the weight changes, food consumption, and survival curve results between male and female mice, we hypothesized that female hormones, such as estradiol, played a multifaceted role which included the regulation of inflammation and immune responses [27]. Research indicated that female hormones had the ability to suppress inflammatory reactions and reduce damage to the intestinal mucosa, thereby contributing to a decreased risk of CRC [28].
In the blood routine tests, ApcL850X mice exhibited significant increases in WBC, MCV, PDW, MPV, NEUT#, and MONO# compared to WT mice. These elevated parameters suggested an inflammatory response occurring within the ApcL850X mice. The decrease in RBC, HGB MCHC, LYMPH%, and EO% suggested concurrent symptoms of anemia in ApcL850X mice. This could possibly be attributed to long-term bleeding from polyps in the gut of ApcL850Xmice [29]. The chronic bleeding from the polyps may lead to a reduction in red blood cells and hemoglobin levels, resulting in anemia. Additionally, the presence of inflammation in the gut may also contribute to the changes in lymphocyte and eosinophil percentages [30].
To ensure the occurrence of the disease, mice aged 22 weeks and above were selected for the analysis of microbiota composition. The analysis of the intestinal microbiota in ApcL850X mice compared to WT mice, between 22 and 26 weeks of age, revealed distinct clustering and significant differences in the composition and proportions of intrinsic bacterial populations. In the intestines of WT mice, a significant proportion of Lactobacillus, a beneficial lactic acid bacterium, was maintained. In contrast, the proportion of Lactobacillus in the intestinal microbiota of ApcL850X mice was significantly reduced, replaced by Alistipes and Bacteroides genera. Studies have shown that Alistipes and Bacteroides genera have pathogenic properties in CRC, and dysbiosis of Bacteroides can promote cancer development due to its negative impact on the host's physiology, metabolism, and immune system, thus facilitating tumor growth. Alistipes and Bacteroides are considered major initiators and promoters of human CRC [20,30].
Furthermore, the combined ROC analysis of gut microbiota composition for CRC prediction supports these findings by quantifying the diagnostic potential of microbial alterations. For female mice, the ROC curve analysis showed an AUC of 0.72 (95% CI: 0.48–0.96), indicating moderate diagnostic accuracy. For male mice, the AUC was 0.67 (95% CI: 0.41–0.93), suggesting slightly lower diagnostic accuracy. These results underscore the potential of gut microbiota composition as a non-invasive diagnostic tool for CRC. However, while the AUC values indicate better-than-random classification, there is still considerable room for improvement.
The difference in AUC values between female and male mice highlights the importance of considering sex-specific factors when developing microbiota-based diagnostic tools. The higher diagnostic accuracy in females suggests that hormonal differences, immune responses, or other sex-specific factors may influence gut microbiota composition and its association with CRC. Future research should explore these sex-specific differences in greater detail to improve diagnostic accuracy and tailor interventions accordingly.
To gain a more comprehensive understanding of these shifts, we conducted a longitudinal analysis tracking microbiota changes from 3 to 25 weeks of age. This analysis revealed dynamic alterations in the relative abundances of key bacterial taxa, including a marked decrease in beneficial bacteria such as Lactobacillus and Bifidobacterium and a concurrent increase in potentially pathogenic taxa like Alistipes and Bacteroides. These longitudinal findings are crucial, as they highlight the temporal dynamics of gut microbiota in relation to CRC development. The progressive loss of beneficial bacteria and the rise in harmful taxa suggest a causal relationship between microbiota dysbiosis and tumorigenesis in the ApcL850X model. By examining these changes over time, our study provides deeper insights into the potential mechanisms by which gut microbiota may influence CRC progression.
In addition to identifying diagnostic biomarkers, the temporal dynamics of gut microbiota changes are also crucial. As demonstrated in earlier analyses (Figure 7a and b), significant shifts in bacterial populations were observed over time, correlating with disease progression. Early detection of such shifts could be pivotal for timely interventions. For example, a decrease in beneficial bacteria like Lactobacillus and Bifidobacterium and an increase in harmful bacteria such as Alistipes might serve as early warning signs of CRC development.
While our study primarily identifies correlations between specific gut microbiota alterations and CRC development in ApcL850X mice, further investigation into the underlying mechanisms is crucial for a deeper understanding of how these microbial changes drive cancer progression [31].
One potential mechanism involves the role of inflammation in CRC. Dysbiosis, characterized by an overrepresentation of pathogenic bacteria such as Alistipes and Bacteroides, has been associated with chronic inflammation in the gut. These bacteria can produce pro-inflammatory molecules, including lipopolysaccharides and other toxins, which may activate immune responses and lead to sustained inflammation. Chronic inflammation is a well-established risk factor for CRC, as it can create a microenvironment conducive to tumorigenesis by promoting cellular proliferation, angiogenesis, and DNA damage [32].
Another mechanism by which gut microbiota may influence CRC progression is through metabolic disruption. Beneficial bacteria like Lactobacillus and Bifidobacterium are known for their roles in producing short-chain fatty acids (SCFAs) such as butyrate, which have anti-inflammatory and anti-carcinogenic properties. The observed reduction in these beneficial taxa in ApcL850X mice could lead to a decrease in SCFA production, weakening the protective barrier of the intestinal mucosa and allowing for increased intestinal permeability. This, in turn, could facilitate the translocation of bacteria and their toxins into the systemic circulation, further promoting inflammation and cancer progression [33].
Moreover, the gut microbiota may impact CRC through immune modulation. The gut microbiota interacts closely with the host’s immune system, influencing the balance between pro-inflammatory and anti-inflammatory responses. The loss of microbial diversity and the shift towards a more pathogenic community structure could impair the immune system’s ability to recognize and eliminate pre-cancerous cells, allowing for unchecked tumor growth [34].
To validate these proposed mechanisms, future studies should focus on exploring the causal relationships between specific bacterial taxa and CRC development. Experimental approaches, such as germ-free mouse models or antibiotic treatment to modulate the gut microbiota, could be used to determine the direct effects of these microbial changes on tumor formation [35]. Additionally, investigating the molecular pathways involved in microbiota-host interactions, including signaling pathways related to inflammation, metabolism, and immune responses, would provide valuable insights into how gut microbiota contribute to CRC pathogenesis.
Additionally, understanding the mechanisms by which gut microbiota influence CRC development is essential. Dysbiosis may contribute to CRC through several pathways, including chronic inflammation, immune modulation, and metabolic disruptions. Alistipes and Bacteroides, for example, are known to produce pro-inflammatory molecules and metabolites that can damage the intestinal mucosa, promote carcinogenesis, and alter immune responses. Unraveling these mechanisms could lead to targeted therapies that restore a healthy microbiota balance, potentially preventing CRC or slowing its progression.
The observed survival rate discrepancies between male and female ApcL850X mice, with all males dying by 23 weeks while 15% of females survived, highlight significant sex-specific differences in CRC progression and overall health outcomes. These differences warrant further investigation to uncover the underlying biological mechanisms and improve the applicability of the ApcL850X mouse model for CRC research.
One possible explanation for the longer survival observed in female mice is the protective role of female hormones, particularly estradiol. Estradiol has been shown to exert anti-inflammatory effects, modulate immune responses, and maintain the integrity of the intestinal mucosa [36]. These properties could help mitigate the inflammatory processes and mucosal damage associated with CRC, leading to slower disease progression and extended survival in female mice.
Additionally, the immune system may respond differently in male and female mice, with females potentially having a more robust or regulated immune response that could better control tumor growth [37]. The interplay between hormones and immune function is a critical area that needs further exploration to understand how these factors contribute to the observed differences in survival rates.
The metabolic differences between males and females could also play a role. Females might metabolize certain nutrients or drugs differently, leading to variations in energy availability and the ability to manage the metabolic demands of a growing tumor [38]. Understanding these metabolic pathways could provide insights into sex-specific vulnerabilities and resilience in CRC.
To fully elucidate the reasons behind these sex-specific survival discrepancies, future studies should consider detailed analyses of hormonal levels, immune profiles, and metabolic markers in both male and female ApcL850X mice. Such investigations could reveal key biological insights that not only explain the differences in survival but also inform the development of sex-specific therapeutic strategies.
Combining gut microbiota analysis with other diagnostic methods could significantly enhance overall diagnostic performance and reliability. For instance, integrating metagenomic data with metabolomic profiles, host genetic information, and traditional clinical markers could provide a more comprehensive and accurate diagnosis. Such multi-modal approaches could leverage the strengths of each method, compensating for their individual limitations.
Addressing these survival discrepancies is crucial for improving the predictive power and translational relevance of the ApcL850X mouse model. By understanding and accounting for these sex-specific differences, researchers can better tailor interventions and improve the model's applicability to human CRC, where similar sex-related differences in disease progression and outcomes have been observed.
While our findings highlight the diagnostic potential of gut microbiota composition in predicting CRC, these results are preliminary and derived from a controlled mouse model. To confirm the robustness and applicability of these findings, broader validation is essential. Future studies should involve larger and more diverse cohorts, including various genetic backgrounds, environmental conditions, and dietary patterns, to ensure the generalizability of the results. Expanding these studies to include human cohorts is particularly important for translating our findings into clinical practice. Human studies could validate the diagnostic biomarkers identified in the ApcL850X mouse model and assess their effectiveness across different populations and stages of CRC.
Translating these findings from the ApcL850X mouse model to human CRC research requires careful consideration of several factors. The mouse model, while valuable, does not fully replicate the complexity of human CRC, which involves a multifaceted interplay of genetic, environmental, and lifestyle factors [39]. Differences in gut microbiota composition between mice and humans, as well as species-specific immune responses and metabolic processes, pose significant challenges in directly applying these results to human patients.
To bridge this gap, it is crucial to undertake comparative studies that evaluate the consistency of gut microbiota biomarkers across species. Integrating data from mouse models with human clinical data through multi-omics approaches, such as metagenomics, metabolomics, and transcriptomics, could enhance our understanding of the shared and divergent pathways involved in CRC progression. Additionally, the development of humanized mouse models, where human microbiota or immune cells are introduced into mice, could provide a more accurate platform for studying the interactions between microbiota and host in a context that more closely resembles human biology [40].
Moreover, the clinical relevance of our findings depends on the ability to implement microbiota-based diagnostics in a clinical setting. This involves addressing practical challenges such as standardizing sample collection and processing, ensuring reproducibility across different laboratories, and developing cost-effective and scalable diagnostic tools. Collaboration with clinical researchers and healthcare providers will be essential to refine these methods and establish protocols that can be seamlessly integrated into existing CRC screening programs.
In summary, while our study offers promising insights into the role of gut microbiota in CRC, broader validation with diverse cohorts, including human studies, is necessary to confirm these findings. Translating these insights into human CRC research and clinical practice will require overcoming significant challenges, but the potential benefits, including more accurate diagnostics and personalized treatments, make this a worthwhile endeavor.
5. CONCLUSION
In conclusion, our findings emphasize the significant role of gut microbiota in CRC development and their potential as diagnostic biomarkers. While the current diagnostic accuracy is moderate, further research into sex-specific differences, temporal dynamics, and multi-modal diagnostic approaches could enhance the utility of gut microbiota analysis. Ultimately, this research paves the way for non-invasive, microbiota-based diagnostics and personalized treatments, improving outcomes for CRC patients.
This study presents a significant advancement in CRC research through the establishment of a spontaneous CRC mouse model with an Apc gene point mutation using CRISPR/Cas9 technology. The comprehensive analyses and innovative approaches employed provide valuable insights into CRC development and the role of gut microbiota. However, addressing the outlined opportunities for improvement, such as enhancing mechanistic insights, ensuring adequate sample sizes, and validating findings in broader contexts, would further solidify the study's contributions to the field.
6. CONFLICTS OF INTEREST
The authors declare that they have no conflicts of interest with the content of the article.
7. FUNDING
This study was supported by the Science and Technology Commission of Shanghai Municipality (No. 2214900102, No. 22DZ2291200), Shanghai Laboratory Animal Research Center "Science and Technology Innovation Plan" Nova project (No.2021NS03, No.2022NS04).
8. AUTHOR CONTRIBUTIONS
All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; agreed to submit to the current journal; gave final approval of the version to be published; and agree to be accountable for all aspects of the work. All the authors are eligible to be an author as per the international committee of medical journal editors (ICMJE) requirements/guidelines.
9. ETHICAL APPROVALS
This study were approved by the Institutional Animal Care and Use Committee of Model Organism Research and Development Department, Shanghai Laboratory Animal Research Center, Shanghai, China (Approval No.: 2211028081) and were conducted in accordance with relevant national and international guidelines.
10. DATA AVAILABILITY
All the data is available with the authors and shall be provided upon request.
11. PUBLISHER’S NOTE
All claims expressed in this article are solely those of the authors and do not necessarily represent those of the publisher, the editors and the reviewers. This journal remains neutral with regard to jurisdictional claims in published institutional affiliation.
12. USE OF ARTIFICIAL INTELLIGENCE (AI)-ASSISTED TECHNOLOGY
The authors declares that they have not used artificial intelligence (AI)-tools for writing and editing of the manuscript, and no images were manipulated using AI.
REFERENCES
1. Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 2021 May;71(3):209–49; CrossRef
2. Fodde R, Smits R, Clevers H. APC, signal transduction and genetic instability in colorectal cancer. Nat Rev Cancer 2001;1:55–67.
3. Mori Y, Nagse H, Ando H, Horii A, Ichii S, Nakatsuru S, et al. Somatic mutations of the APC gene in colorectal tumors: mutation cluster region in the APC gene. Hum Mol Genet 1992;1:229–33.
4. Ren J, Sui H, Fang F, Li Q, Li B. The application of Apc(Min/+) mouse model in colorectal tumor researches. J Cancer Res Clin Oncol 2019;145(5):1111–22.
5. Groden J, Thliveris A, Samowitz W, Carlson M, Gelbert L, Albertsen H, et al. Identification and characterization of the familial adenomatous polyposis coli gene. Cell 1991;66:589–600.
6. Galiatsatos P, Foulkes WD. Familial adenomatous polyposis. Am J Gastroenterol 2006;101:385–98.
7. Logan CY, Nusse R. The Wnt signaling pathway in development and disease. Annu Rev Cell Dev Biol 2004;20:781–810.
8. Schatoff EM, Leach BI, Dow LE. Wnt signaling and colorectal cancer. Curr Colorectal Cancer Rep 2017;13:101–10.
9. Morin PJ, Sparks AB, Korinek V, Barker N, Clevers H, Vogelstein B, et al. Activation of beta-catenin-Tcf signaling in colon cancer by mutations in beta-catenin or APC. Science 1997 Mar 21;275(5307):1787–90.
10. van Neerven SM, de Groot NE, Nijman LE, Scicluna BP, van Driel MS, Lecca MC, et al. Apc-mutant cells act as supercompetitors in intestinal tumour initiation. Nature 2021;594(7863):436–41.
11. Brocardo M, Lei Y, Tighe A, Taylor SS, Mok MTS, Henderson BR. Mitochondrial targeting of adenomatous polyposis coli protein is stimulated by truncating cancer mutations: regulation of Bcl-2 and implications for cell survival. J Biol Chem 2008;283(9):5950–9.
12. Bodmer WF, Bailey CJ, Bodmer J, Bussey HJ, Ellis A, Gorman P, et al. Localization of the gene for familial adenomatous polyposis on chromosome 5. Nature (London) 1987;328:614–6.
13. Rowan AJ, Lamlum H, Ilyas M, Wheeler J, Straub J, Papadopoulou A, et al. APC mutations in sporadic colorectal tumors: a mutational "hotspot" and interdependence of the "two hits". Proc Nat Acad Sci 2000;97(7):3352–7.
14. Becker WR, Nevins SA, Chen DC, Chiu R, Horning AM, Guha TK, et al. Single-cell analyses define a continuum of cell state and composition changes in the malignant transformation of polyps to colorectal cancer. Nat Genet 2022;54(7):985–95.
15. Half E, Bercovich D, Rozen P. Familial adenomatous polyposis. Orphanet J Rare Dis 2009;4:1–23.
16. Cheng Y, Ling Z, Li L. The intestinal microbiota and colorectal cancer. Front Immunol 2020;11(1):615056.
17. Wong SH, Yu J. Gut microbiota in colorectal cancer: mechanisms of action and clinical applications. Nat Rev Gastroenterol Hepatol 2019;16(3):1–15.
18. Söderlund S, Brandt L, Lapidus A, Karlén P, Broström O, Löfberg R, et al. Decreasing time-trends of colorectal cancer in a large cohort of patients with inflammatory bowel disease. Gastroenterology 2009;136(5):1561–7.
19. Moser AR, Pitot HC, Dove WF. A dominant mutation that predisposes to multiple intestinal neoplasia in the mouse. Science 1990;247(4940):322–4.
20. Turkington CJR, Varadan AC, Grenier SF, Grasis JA. The viral Janus: viruses as aetiological agents and treatment options in colorectal cancer. Front Cell Infect Microbiol 2021;10:601573.
21. Jackstadt R, Sansom OJ. Mouse models of intestinal cancer. Comp Med 2016;238(2):141–51.
22. Chen S, Zhou Y, Chen Y, Gu J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018;34(17):i884–90; CrossRef
23. Mago? T, Salzberg SL. FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics 2011;27(21):2957–63; doi:10.1093/bioinformatics/btr507
24. Edgar RC. UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat Methods 2013;10(10):996–8; CrossRef
25. Stackebrandt E, Goebel BM. Taxonomic note: a place for DNA-DNA reassociation and 16S rRNA sequence analysis in the present species definition in bacteriology. Int J Syst Bacteriol 1994;44(4):846–9; doi:10.1099/00207713-44-4-846
26. Wang Q, Garrity GM, Tiedje JM, Cole JR. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol 2007;73(16):5261–7; doi:10.1128/AEM.00062-07
27. DeCosse JJ, Ngoi SS, Jacobson JS, Cennerazzo WJ. Gender and colorectal cancer. Eur J Cancer Prev 1993;2(2):105–15.
28. Lin JH, Zhang SM, Rexrode KM, Manson JE, Chan AT, Wu K, et al. Association between sex hormones and colorectal cancer risk in men and women. Clin Gastroenterol Hepatol 2013;11(4):419–24.
29. Chao AC, Chan JS, Cheung VC, et al. Tu1088 an epidemiological study on incidence of hospitalization and in-hospital mortality of peptic ulcer bleeding, gastrointestinal neoplasms, and non-infectious enterocolitis: a population-based study from 2005 to 2014. J Gastroenterol 2016;150(4):S838.CrossRef
30. Torres-Maravilla E, Boucard AS, Mohseni AH, Taghinezhad-S S, Cortes-Perez NG, Bermúdez-Humarán LG. Role of gut microbiota and probiotics in colorectal cancer: onset and progression. Microorganisms 2021;9(5):1021.
31. Kostic AD, Chun E, Robertson L, Glickman JN, Gallini CA, Michaud M, et al. Fusobacterium nucleatum potentiates intestinal tumorigenesis and modulates the tumor-immune microenvironment. Cell Host Microbe 2013 Aug 14;14(2):207–15; CrossRef
32. Arthur JC, Perez-Chanona E, Mühlbauer M, Tomkovich S, Uronis JM, Fan TJ, et al. Intestinal inflammation targets cancer-inducing activity of the microbiota. Science 2012 Oct 5;338(6103):120–3; CrossRef
33. Donohoe DR, Garge N, Zhang X, Sun W, O'Connell TM, Bunger MK, et al. The microbiome and butyrate regulate energy metabolism and autophagy in the mammalian colon. Cell Metab 2011 May 4;13(5):517–26; CrossRef
34. Atarashi K, Tanoue T, Oshima K, Suda W, Nagano Y, Nishikawa H, et al. Treg induction by a rationally selected mixture of Clostridia strains from the human microbiota. Nature 2013 Aug 8;500(7461):232–6; CrossRef
35. Gopalakrishnan V, Spencer CN, Nezi L, Reuben A, Andrews MC, Karpinets TV, et al. Gut microbiome modulates response to anti-PD-1 immunotherapy in melanoma patients. Science 2018 Jan 5;359(6371):97–103; CrossRef
36. Lin JH, Zhang SM, Rexrode KM, Manson JE, Chan AT, Wu K, et al. Association between sex hormones and colorectal cancer risk in men and women. Clin Gastroenterol Hepatol 2013 Apr;11(4):419–24.e1; CrossRef
37. Klein SL, Flanagan KL. Sex differences in immune responses. Nat Rev Immunol 2016 Oct;16(10):626–38; CrossRef
38. Mauvais-Jarvis F. Sex differences in metabolic homeostasis, diabetes, and obesity. Biol Sex Differ 2015 Sep 3;6:14; CrossRef
39. Seok J, Warren HS, Cuenca AG, Mindrinos MN, Baker HV, Xu W, et al. Genomic responses in mouse models poorly mimic human inflammatory diseases. Proc Natl Acad Sci U S A 2013 Feb 26;110(9):3507–12; CrossRef
40. Shultz LD, Ishikawa F, Greiner DL. Humanized mice in translational biomedical research. Nat Rev Immunol 2007 Feb;7(2):118–30; CrossRef