1. INTRODUCTION
Sesame (Sesamum indicum L., 2n = 26), from the Pedaliaceae family, is a genetically rich crop comprising 73 species [1]. With the increase in global population, sesame consumption has exponentially increased to meet its nutritional value, including protein, oils, and carbohydrates [2]. It also has pharmacological importance due to sesamin and sesamolin, which have antioxidative, anti-inflammatory, antihypertensive, and anticarcinogenic properties [3]. However, the production of sesame seeds was lesser in 2022 (0.71 million metric tons) compared with 2021 [4]. Therefore, a comprehensive analytical review is required to study its genetic richness and health-linked nutritional components, identified genes/QTLs for various biotic and abiotic stresses, and quality traits [5].
Besides having nutritional value, sesame is highly rich in polysaccharides, including mucilage, starch, cellulose, and pectin. Among them, cellulose is present at ample concentration and comprises 47% of the total polysaccharide in a hull of sesame seeds [6]. Moreover, significant amounts of secondary metabolites, such as polyphenols, flavonoids, and lignin, are present in sesame crops. These polysaccharide components and secondary metabolites restricted DNA extraction with an intact quality, yield, and utility in molecular studies [7]. In addition, these biomolecules have a structural resemblance to the nucleic acids, leading to hindrances in purified DNA extraction [8].
DNA extraction is crucial for studying genomes in molecular biology and plant breeding. Using purified DNA, numerous molecular studies can be performed from basic molecular to the complex level, including gene amplification and variations within the genome, gene tagging, traits linked to the gene of interest, genomewide association study, and high-throughput genome sequencing, like next-generation technologies. In addition, DNA conservation and storage are robustly increased to understand the phylogenetic and evolutionary relationship between the plants [9]. However, achieving the purified DNA takes time to perform or conduct the desired study because of the richness of phenolic and polysaccharide components in plant-based tissue systems. Therefore, high-quality purified DNA must be isolated from the plant tissue to eliminate such hindrances.
Multiple methods like SDS, cetyl-tetra methyl ammonium bromide (CTAB), and commercially available kits have been formulated to get the DNA with high purity from plants. However, using a kit for many samples for DNA extraction is very expensive regarding the cost per-sample ratio, for example; QIAGEN (DNeasy Plant Pro Kit) and the genomic DNA (gDNA) extraction will cost nearly 600 Indian rupees per sample. The classical DNA extraction method solely depends on a large amount of tissue (usually in grams) directly associated with plant growth [10,11]. The reagent preparation takes 3–4 h, including autoclave. Besides, it is cost-effective and the cost for each sample ranges from 8 to 10 Indian rupees. Therefore, classical methods allow the researcher to extract the bulk samples in a cost-independent manner. However, time consumption is the principal disadvantage of these protocols as the large quantity of tissue cannot be recovered quickly. Commercial kits take 3–4 h, while the proposed protocol takes 4 days to complete the DNA extraction procedure. However, commercial kits do not meet the concentration requirement in sesame crop but CTAB 2.5× protocol showed sufficient concentration that can be used for downstream application.
The primary objective of this research work is to standardize the DNA extraction protocol from the molecular complex matrix of the cells of sesame plant. To accelerate this investigation, three different methods were employed in sesame roots and leaf tissue to standardize the DNA extraction protocol. Moreover, we also focused on and compared it to check the quantity of DNA in two ways: (a) DNA extraction in combination with purification and (b) directly adding RNase into the CTAB buffer. To the best of our knowledge, no such report has been available to determine the DNA quality and quantity in different sesame tissues (root and shoot). Hence, the study presents a quick, cost-effective, and less laborious method to extract the DNA, following standard laboratory conditions.
2. MATERIALS AND METHODS
2.1. Plant Material
Seeds of four sesame accessions (IC129289, EC377024, PB-Til 2, and Shwetha) were collected from the National Bureau of Plant Genetic Resources, New Delhi, and allowed to germinate in soil-rite till the seedling stage (15–20 days). The plant growth and maintenance were monitored at the phenomics facility in controlled conditions at a temperature of 28 ± 3°C, 55–60% relative humidity, and 14 and 8 h of light and dark photoperiod [Figure 1]. Root and shoot (fully opened leaf with stem) tissue samples were harvested and stored at −80°C for gDNA extraction.
 | Figure 1: Four genotypes at seedling stage were grown for DNA extraction. Seedlings were allowed to grow till 15 days after germination. Leaf and root tissues were harvested and stored at −80°C for further use in DNA extraction.
[Click here to view] |
2.2. Reagents
Chemicals and reagents were procured from Merck to conduct the present study. Autoclaved Millipore water from the Millipore Milli-RO4 reverse osmosis system was used to prepare the gDNA extraction solutions.
The details of the reagents used to prepare the solutions are as follows: CTAB, sodium chloride, ethylenediamine tetra-acetic acid (EDTA), polyvinylpyrrolidone (PVP–), Tris-chloride, 2-mercaptoethanol, chloroform:isoamyl alcohol (24:1), phenol:chloroform:isoamyl alcohol (25:24:1), Tris- EDTA, ethanol, RNase A, chloroform, sodium acetate, and nuclease-free water.
2.3. Extraction Buffers
The methodology to obtain pure DNA without RNA and polysaccharide contamination is described in Figure 2. Three different DNA extraction buffers (DEBs) were used for the shoot and root tissues to standardize the protocol for gDNA extraction from sesame.
 | Figure 2: The sesame seedlings were subjected to DNA extraction following three different methods. (A) Doyle and Doyle method, which represents the difficulty to eliminate RNA contamination. (B) CTAB 2.5× buffer demonstrates high-quality DNA. (C) Modified CTAB 2.5× buffer represents low-quality DNA, RNA, and polysaccharide contamination.
[Click here to view] |
1. CTAB 2.5× Buffer
1.5 M sodium chloride (NaCl), 1% PVP, 25 EDTA, 100 Tris-Cl (8.8 pH), 0.2% β-mercaptoethanol, and 2.5% CTAB.
2. Doyle and Doyle Method
100 mM Tris-chloride with a pH of 8.0, 20 mM EDTA, 2% CTAB, 1.4 M NaCl, and 0.2% β-mercaptoethanol.
3. Modified CTAB 2.5× Buffer
The buffer preparation is the same as the CTAB 2.5× buffer, but a few steps were modified to reduce the time consumption.
2.4. DNA Extraction Methodology – Protocol 1 (CTAB 2.5× Buffer)
This is divided into three steps: (a) cell lysis, (b) protein precipitation, and (c) purification. This protocol is an adaptation of the CTAB DNA extraction method [12], which was carried out to extract strawberry DNA. With several modifications in solution preparation and concentration, the adapted protocol was used in sesame crops to keep gDNA intact with high purity.
a) Extraction
Cell or tissue lysis is the first stage to achieve the pure and integrated DNA. To perform the cellular lysis, a fresh solution of CTAB 2.5× buffer containing 1.5 M sodium chloride (NaCl), 1% PVP, 25 mM EDTA, 100 mM Tris-Cl (pH 8.8), 0.2% β-mercaptoethanol, and 2.5% CTAB was prepared. Harvested shoot and root tissue of approximately 100 g were crushed in liquid nitrogen into a fine powder using a prechilled mortar and pestle. Meanwhile, the solution was allowed to preheat in a water bath at 65°C for 30 min. Adding the CTAB buffer to the pulverized tissue will enable the buffer solution to attain room temperature (RT). Pulverized tissue was placed into 2 ml polypropylene tubes (Eppendorf) containing 1 ml of preheated DEB following suspension incubation at 65°C for 1 h with intermittent mixing by gentle swirling.
b) Protein Precipitation
After cellular lysis, protein precipitation was performed by adding an equal volume of chloroform:isoamyl alcohol (24:1), and the samples were gently mixed to avoid shearing. Then, the samples were centrifuged at 9,464 g for 15 min at RT to separate the organic and aqueous phases. The aqueous phase was carefully collected and transferred into a fresh tube of 1.5 ml. To the aqueous phase, 650 µl of prechilled isopropanol solution was added, followed by gently mixing and inverting the tubes for 30 s, which leads to DNA precipitation by dehydrating the surrounding environment of DNA from the aqueous phase. The sample matrix was then stored at −20°C for overnight incubation. The solution mixture was centrifuged at 13,000 RPM for 20 min, and the supernatant was discarded. The precipitated DNA was washed twice with 70% chilled ethanol and centrifuged at 2744 g for 7 min. Finally, the pellet was dissolved in 200 µl Tris-EDTA (TE) buffer (pH 8.0). The samples were incubated overnight at 4°C to dissolve the pellet uniformly.
c) Purification
To achieve the purified DNA, 3 µl of RNase A solution (10 mg/µl) was added to the pellet and incubated at 37°C for 30 min. In the dissolved pellet (nonpurified), an equal volume of phenol:chloroform:isoamyl-alcohol (25:24:1) mixture was added and gently mixed by inverting the tubes, which will accelerate the purification process by forming a complex with the remaining lipids and polysaccharides. The solution mixture containing the DNA in an Eppendorf tube was centrifuged at 8,750 g for 20 min at 4°C. The aqueous phases were collected in a fresh 1.5 ml tube. To the collected aqueous phase, an equal volume of chloroform was added and centrifuged at 8,064 g for 15 min at 4°C, and the aqueous phase was collected. Besides, 1/10 volume of 3 M sodium acetate was added, and the solution was gently mixed. The Eppendorf tubes were then filled with 100% chilled ethanol, followed by overnight incubation at −20°C. After incubation, the tube containing precipitated DNA was centrifuged at 5,600 g at 4°C for 30 min. Finally, the supernatant was discarded, and the pellet was washed with 70% ethanol and centrifuged at 5,054 g for 7 min. The collected pellets were air-dried, and 100 µl of TE buffer was added to suspend the pellet. The extracted DNA samples were stored at 4°C to dissolve the pellet.
2.5. Protocol 2 (Doyle and Doyle Method)
The present protocol is a CTAB-based method [13] widely accepted for DNA extraction in rice and maize crops [14,15]. The present study prepared a fresh solution of DEB comprising 100 mM Tris-chloride, pH 8.0, 2% CTAB, 20 mM EDTA, 1.4 M NaCl, and 0.2% β-mercaptoethanol. The solution was allowed to be preheated at 60°C for 30 min before DNA extraction. Approximately 150 mg of tissues were weighed and crushed into fine powder in liquid nitrogen using a prechilled mortar and pestle. The pulverized tissue was placed into 2 ml tubes (Eppendorf) containing 580 µl of preheated DEB following incubation at 60°C for 1 h with intermittent mixing by gentle swirling. Then, an equal volume of chloroform:isoamyl alcohol (24:1) was added, and the samples were mixed gently to avoid DNA shearing and damage. The lyophilized matrix solutions were centrifuged at 5,600 g at RT for 15 min. The aqueous phase was then collected with the help of a 1 ml pipette and transferred into a fresh tube of 1.5 ml. To avoid RNA contamination, 2 µl of RNase A solution (10 mg/µl) was added to the aqueous phase, and the solution was kept at 37°C for 30 min. The samples were collected from the heat blocker, and chloroform:isoamyl alcohol (24:1) was added (equal volume), followed by gentle mixing and centrifugation at 10,000 RPM at RT for 10 min. The supernatant was collected in a fresh 1.5 ml tube. Ice-cold isopropanol was added to the 0.6 volume of the supernatant and incubated at −20°C for 30 min. The matrix mixture was centrifuged at 5,600 g at 4°C for 10 min. The sedimented pellet was taken and washed with prechilled 70% ethanol. Finally, the pellet was air-dried and dissolved in 100 µl of nuclease-free water.
2.6. Protocol 3 (Modified CTAB 2.5× Buffer Method)
Here, the CTAB protocol (Protocol 1) was modified to reduce the time consumption and get highly purified DNA. In this method, a few steps were removed from Protocol 1. Buffer preparation was the same as described in the CTAB 2.5× buffer method. Furthermore, shoot and root tissues (100 mg) were collected and crushed into fine powder in liquid nitrogen using a prechilled mortar and pestle. The pulverized tissue was placed into 2 ml tubes (Eppendorf) containing 1 ml of preheated DEB and 3 µl of RNase A solution (10 mg/µl) following incubation at 65°C for 1 h with intermittent mixing by gentle swirling. The samples were allowed to reach RT, and once the mixture attained RT, an equal volume of chloroform:isoamyl alcohol (24:1) was added and centrifuged at 9,464 g at RT for 15 min. The supernatant was collected and transferred to a fresh 1.5 ml tube. Then, 650 µl of prechilled isopropanol solution was added with 1/10th of 3 M sodium acetate. The samples were then stored at −20°C for overnight incubation. The solution mixture was then centrifuged at 9,464 g for 20 min. The supernatant was discarded, and the pellet was washed twice with 70% chilled ethanol, followed by centrifugation at 2,744 g for 7 min.
2.7. DNA Quantification
Plant gDNA samples were run on a 0.8% agarose gel at 80 V for 30 min to determine the DNA quality. The DNA was quantified with the help of nanodrop (Thermo Fisher Scientific, NanoDrop One C), and the purity was determined by measuring the absorbance at 260 and 280 nm.
3. RESULTS AND DISCUSSION
The gDNA extraction from plants with high purity and integrity is essential to carry out further molecular analysis, including polymerase chain reaction, marker-assisted selection, and next-generation sequencing. Therefore, purification is crucial as it eliminates RNA contamination and the remains of phenolics and polysaccharide components. To follow the standardization method of DNA to a polysaccharide-rich crop, DNA extraction was preceded by the young leaves of the seedling stage of the four genotypes of the S. indicum. A wide range of DNA concentrations was recorded [Table 1] while employing all three DNA extraction methods.
Table 1: Stock and working concentration of CTAB buffer.
| Chemicals | Stock Concentration | Working Concentration | Remark |
|---|
| Tris-chloride | 1 M | 100 mM | It is essential to maintain pH 8.0 |
| Ethylenediamine tetraacetic acid (EDTA) | 0.5 M | 20 mM | It should be dissolved completely, and maintaining pH 8.0 is compulsory |
| Sodium chloride (NaCl) | 5M | 1.5 M | Complete dissolution is a prerequisite |
| Cetyltrimethylammonium bromide (CTAB) | 10% | 2.5% | For the proper dissolution, it is required to keep CTAB at 60°C |
| Polyvinylpyrrolidone (PVP) | | 1% | Always it is required to weigh fresh PVP and dissolve it in distilled water |
The ratio of DNA (A260 and A280) varied in all three DNA extraction methods. Doyle and Doyle method depicts the DNA concentration in the shoot varied from 201 to 680 ng/µl and purity from 1.4 to 1.68 in four genotypes. Whereas in the root tissues, its concentration varied from 185 to 388 ng/µl, and purity was around 1.6–1.72. In the CTAB 2.5× method, the concentration of DNA varied from 273 to 383 ng/µl, and 266–640 ng/µl in the leaf and root, respectively, and the purity ranged from 1.8 to 1.9 in both the tissues of the four genotypes. Compared to Doyle and Doyle method, the modified CTAB method showed DNA purity of 1.44–1.60 in shoot and 1.50–1.61 in root, whereas the concentration varied from 34.2 to 876 ng/µl and from 110 to 446 ng/µl in leaf and root tissues, respectively. The purity of DNA depends on the absorbance ratio of 260/280, and a contamination-free DNA (devoid of protein and phenolic content) always represents a ratio of 1.8 [16]. In addition, an absorption ratio of more than 2 depicted the acetone or alcohol residue while performing DNA extraction or purification [17].
The present study employed different methods to resolve the polysaccharide-related issues that barricade to isolate the purified DNA. However, the CTAB-based method with a high salt concentration represents a promising result for DNA extraction with high purity and intact quality. The exact composition of the lysis buffer is a limiting factor influencing the DNA yield. Most of the reports need to indicate the buffer composition, leading to difficulties in troubleshooting. Here, the described buffer composition [Table 2] will help breeders and researchers to conduct their genetic investigation, which is directly associated with the high quality of DNA in the oilseeds crop. Moreover, Protocol 1 is an adaptation of the methodology described by Porebski et al. [12] and is different in several ways. The present protocol relies on a high concentration of CTAB (2.5%) and PVP (1%). Here, chloroform was used in combination with isoamyl alcohol (which is a butanol) but in the previous protocol octanol was used. The increased centrifugation time improved the separation of aqueous phase from the organic phase, which was a limitation in the referred protocol. In addition, phenol:chloroform:isomamyl alcohol when used in combination was found better in terms of eliminating the polysaccharide contaminations. Finally, incubation and cold storage time with isopropanol were increased for the improved precipitation of DNA, which was done by using ethanol in the refereed protocol [12]. The CTAB 2.5× method incorporated a few steps to extract the DNA with improved quality and yield. The cell membrane of leaf tissue was disrupted by using liquid nitrogen. Liquid nitrogen helps deactivate the cellular enzymes by implying the heat energy generated while crushing the tissues to reduce the probability of DNA damage and shearing [18].
Table 2: The concentration of DNA was extracted from three methods with 260/280 and 260/230 ratios to see the impurity of protein and RNA. The values are presented with mean ± standard deviation.
| Methods | CTAB 2.5× Buffer | Doyle and Doyle Method | Modified CTAB 2.5× Buffer |
|---|
| | Ratio | | Ratio | | Ratio |
|---|
| Genotypes | DNA Concentration (ng/µl) | 260
/280 | 260
/230 | DNA Concentration (ng/µl) | 260
/280 | 260
/230 | DNA Concentration (ng/µl) | 260
/280 | 260
/230 |
|---|
| Shoot |
| IC129289 | 383±21 | 1.84 | 1.89 | 680.1±62 | 1.52 | 2.38 | 125±28 | 1.58 | 2.11 |
| EC377024 | 345.9±34 | 1.8 | 1.85 | 721±65 | 1.47 | 2.41 | 876±42 | 1.59 | 2.34 |
| PB-Til 2 | 273.1±19 | 1.87 | 1.9 | 110±24 | 1.68 | 2.22 | 784.4±51 | 1.44 | 2.3 |
| Shwetha | 297.4±45 | 1.79 | 1.84 | 201±27 | 1.64 | 2.19 | 34.2±13 | 1.6 | 1.98 |
| Root |
| IC129289 | 266±63 | 1.85 | 1.87 | 388±43 | 1.6 | 2.17 | 421.8±36 | 1.5 | 2.22 |
| EC377024 | 472.4±49 | 1.88 | 1.94 | 185.6±18 | 1.61 | 2.2 | 446±35 | 1.55 | 2.28 |
| PB-Til 2 | 584.6±58 | 1.92 | 1.91 | 192.7±32 | 1.69 | 2.29 | 110.5±9 | 1.61 | 2.29 |
| Shwetha | 640±51 | 1.8 | 1.88 | 250.2±28 | 1.72 | 1.98 | 173±35 | 1.54 | 2.2 |
Concentrated 3× CTAB is generally used to enhance the sufficient lysis of cells and the nuclear membrane. The genetic component remains exposed from the cellular matrix [19]. The present method used 2.5× CTAB with a 0.2% concentration of β-mercaptoethanol to eliminate the polyphenols. In addition, 0.3% of β-mercaptoethanol was used, followed by Li et al. [20]. Apart from containing β-mercaptoethanol, the DEB or lysis buffer contains 1.4 M NaCl to enhance the DNA quality [8]. The CTAB 2.5× buffer method was used with 1.5 M NaCl solution. The added salt significantly removed impurities like polysaccharides and produced high-quality DNA. Variability among the DNA extraction methods indicates the varied concentration of buffer composition and different chemicals used to precipitate and purify the DNA [21].
The altered buffer compositions affected the DNA purity and quality. The plausible cause behind this comprised several factors like structural, biochemical, and genetic alteration between the leaf tissues of plant species and treatment duration with a varied concentration of a particular chemical [22]. Therefore, it is a prerequisite to standardize the DNA extraction protocol before initiating any molecular discovery at the genetic level because plant species have a diverse range of secondary metabolites, which interfere with DNA purity and yield [23]. Leaf selection was also critical for DNA extraction for oil seed crops. The green and fully opened young leaf tissues could provide a good yield of DNA. Moreover, dried and overmature leaves are obstacles in DNA extraction by increasing the chances of contamination with phenols or secondary metabolites [12]. Therefore, fully opened young leaf tissue is suitable for DNA extraction.
In addition to the choice of tissue selection and buffer composition, cellular debris, proteins, and lipids were selectively eliminated by forming the complex with organic compounds. These impurities were removed using chloroform:isoamyl alcohol (24:1). Complete DNA precipitation was crucial in gDNA extraction. DNA precipitation is the most critical step in a sesame crop due to its richness in polyphenols, proteins, and lipids [24]. Therefore, the extended incubation period (overnight) with chilled isopropanol at – 20°C favored the DNA precipitation as reported by Michiels et al. [25], where they mentioned that DNA quality and quantity depend on two factors: duration of the DNA precipitation and temperature.
To assess DNA yield, centrifugation timing has been increased here for proper sedimentation of an organic compound and efficient separation of the aqueous layer. Furthermore, the cellular extract should be treated with RNase A to remove the RNA contamination [26]. After extraction, the DNA pellet was diluted in 1× TE buffer for better stability. It is worth noting that when free from contamination the DNA pellet is easier to dissolve either in the TE buffer or in the nuclease-free water [27]. While extracting the DNA, it was noticeable that RNA contamination persists in the leaf and root tissue of sesame, followed by Doyle and Doyle and the modified CTAB 2.5× buffer method [Figure 2]. However, no contamination was observed in the CTAB 2.5× buffer method.
Besides the CTAB 2.5× buffer method, Doyle and Doyle and the modified CTAB 2.5× method did not demonstrate a promising result. Both methods represent DNA with high impurities of RNA and phenolic content. The possible cause behind this issue is the variation in buffer composition and time employed to extract the DNA. Moreover, our method is slightly time-consuming and serves the DNA with many qualities and yields. Therefore, the CTAB 2.5× buffer method experienced a suitable advantage over the remaining two methods, which have been undertaken to standardize the DNA extraction protocol in oilseed crops. Besides, this method can be used to assess the waterlogging responses [28], genotype characterization, and QTLs mapping. Using this method, we extracted DNA from 1200 genotypes of sesame [Figure 3] to be sequenced for genotyping, and all of them were passed for sequencing.
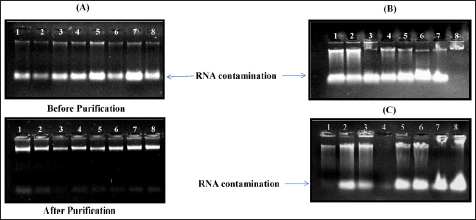 | Figure 3: Three different methods for DNA extraction for four genotypes: (A) the CTAB 2.5× buffer method, (B) Doyle and Doyle method, and (C) the modified version of the CTAB 2.5× method. All three methods were utilized to extract the genomic DNA from four different genotypes of sesame. DNA was extracted from the shoot (lanes 1–4) and the root tissues (lanes 5–8). High-quality DNA with sharp bands was obtained using the CTAB 2.5× method, whereas RNA and polyphenolic contamination can be seen in (B) and (C).
[Click here to view] |
4. CONCLUSION
The present study demonstrated a quicker, more accessible way to extract the gDNA from the polysaccharide and polyphenols-rich crops. Sesame falls into these categories, and extracting the DNA with high purity was a great challenge. DNA precipitation and mucilage were the major challenges to extract the pure DNA. The presence of mucilage triggers the free radical’s generation, which when reacting with cellular molecules and oxygen causes significant DNA damages and yield loss. Therefore, DNA precipitation was done using a high salt concentration that barricade the mucilage interference to DNA. Moreover, DNA was incubated for longer duration to selectively eliminate the mucilage and polysaccharide contamination. In the present study, three different methods were utilized to extract the DNA, and the CTAB 2.5× buffer method illustrated a robust method to extract DNA in its organized form. However, this method is somewhat time-consuming. Still, reducing the extended time is unavoidable as more extended and cold precipitation enhances the DNA purity and quality by sufficiently eliminating the phenolic debris. While Doyle and Doyle and the modified CTAB 2.5× method represent few promising results, both failed to remove RNA and phenolic contamination altogether. Moreover, the CTAB 2.5× buffer method is cost-effective and a potential method to extract DNA across the corners of scientific laboratories or countries. In addition, the presented protocol may play an essential role in detecting food concern safety, conservation of biodiversity, and identification and validation of genetically modified molecular breeding approaches. Therefore, using the CTAB 2.5× method, there are tremendous opportunities to detect economically related traits, including quantitative and qualitative. However, using genome sequencing and QTL mapping, it may participate in water-logging and drought tolerance. In addition, resistant or tolerant genes can be incorporated into susceptible varieties using molecular breeding techniques to secure the nutritional requirements.
5. ACKNOWLEDGMENTS
The authors acknowledge the funding support by the Department of Biotechnology (DBT), New Delhi, India (no. BT/Ag/Network/Sesame/2019-20).
6. AUTHOR CONTRIBUTIONS
VR conceptualized and designed the work. AS, NG, NJ, and PM were involved in the experimentation. VR and AS drafted the manuscript. SK, RK, and NKS finalized and edited the whole manuscript.
7. CONFLICTS OF INTEREST
The authors report no financial or any other conflicts of interest in this work.
8. ETHICAL APPROVALS
The project, in its meticulous design and execution, has been structured to not require any ethical approval, a testament to its adherence to the highest ethical standards.
9. DATA AVAILABILITY
All the data is available with the authors and shall be provided upon request.
10. USE OF ARTIFICIAL INTELLIGENCE (AI)-ASSISTED TECHNOLOGY
The authors confirm that there was no use of artificial intelligence (AI)-assisted technology for assisting in the writing or editing of the manuscript and no images were manipulated using AI.
11. PUBLISHER’S NOTE
All claims expressed in this article are solely those of the authors and do not necessarily represent those of the publisher, the editors and the reviewers. This journal remains neutral with regard to jurisdictional claims in published institutional affiliation.
REFERENCES
1. WFO. World Flora Online 2023. 2023. Available from: http://www.worldfloraonline.org (November 24, 2023).
2. Myint D, Gilani SA, Kawase M, Watanabe KN. Sustainable sesame (Sesamum indicum L.) production through improved technology: an overview of production, challenges and opportunities in Myanmar. Sustainability. 2020;12:3515. [CrossRef]
3. Dossou SSK, Xu, FT, Dossa K, Rong Z, Zhao YZ, Wang, LH. Antioxidant lignans sesamin and sesamolin in sesame (Sesamum indicum L.): a comprehensive review and future prospects. J Integr Agricult. 2023;22:14–30. [CrossRef]
4. Statista Research Department. Production volume of sesame across India from financial year 2013 to 2022, with estimates until 2023. Statista Research Department. Statista; 2023.
5. Yadav R, Kalia S, Rangan P, Pradheep K, Rao GP, Kaur V, et al. Current research trends and prospects for yield and quality improvement in sesame, an important oilseed crop. Front Plant Sci. 2022;13:863521. [CrossRef]
6. Yao YG, Wang WY, Chen LY, Liu HM, Yan RZ, Li S, et al. Structural changes of cellulosic polysaccharides in sesame hull during roasting at various temperatures. Qual Assur Saf. 2021;13:13–24. [CrossRef]
7. Khanuja SPS, Shasany AK, Darokar MP, Kumar S. Rapid isolation of DNA from dry and fresh samples of plants producing large amounts of secondary metabolites and essential oils. Plant Mol Biol Rep. 1999;17:74. [CrossRef]
8. Sahu SK, Thangaraj M, Kathiresan K. DNA extraction protocol for plants with high levels of secondary metabolites and polysaccharides without using liquid nitrogen and phenol. ISRN Mol Biol. 2012;1–6. [CrossRef]
9. Dharajiya DT, Shah A, Galvadiya BP, Patel MP, Srivastava R, Pagi NK, et al. Genome-wide microsatellite markers in castor (Ricinus communis L.): identification, development, characterization, and transferability in Euphorbiaceae. Ind Crops Prod. 2020;151:112461. [CrossRef]
10. Jobes DV, Hurley DL, Thien LB. Plant DNA isolation: a method to efficiently remove polyphenolics, polysaccharides, and RNA. Taxon. 1995;44:379–86. [CrossRef]
11. Cheng YJ, Guo WW, Yi HL, Pang XM, Deng X. An efficient protocol for genomic DNA extraction from Citrus species. Plant Mol Biol Rep. 2003;21:177–8. [CrossRef]
12. Porebski S, Bailey LG, Baum BR. Modification of a CTAB DNA extraction protocol for plants containing high polysaccharide and polyphenol components. Plant Mol Biol Rep. 1997;15:8–15. [CrossRef]
13. Doyle J. DNA protocols for plants. Mol Tech Taxonomy. 1991;57:283–93. [CrossRef]
14. Ying Y, Hu Y, Zhang Y, Tappiban P, Zhang Z, Dai G, et al. Identification of a new allele of soluble starch synthase IIIa involved in the elongation of amylopectin long chains in a chalky rice mutant. Plant Sci. 2023;328:111567.
15. Kamara MM, Ghazy, NA, Mansour E, Elsharkawy MM, Kheir AM, Ibrahim KM. Molecular genetic diversity and line× tester analysis for resistance to late wilt disease and grain yield in maize. Agronomy 2021;11:898. [CrossRef]
16. Abdel-Latif A, Osman G. Comparison of three genomic DNA extraction methods to obtain high-quality DNA from maize. Plant Methods. 2017;13:1–9. [CrossRef]
17. Webb DM, Knapp SJ. DNA extraction from a previously recalcitrant plant genus. Plant Mol Biol Rep. 1990;8:180–5. [CrossRef]
18. Aboul-Maaty NAF, Oraby HAS. Extraction of high-quality genomic DNA from different plant orders applying a modified CTAB-based method. Bull Natl Res Cent. 2019;43:1–10. [CrossRef]
19. Amani J, Kazemi R, Abbasi AR, Salmanian AH. A simple and rapid leaf genomic DNA extraction method for polymerase chain reaction analysis. Iran J Biotechnol. 2011;9:69–71.
20. Li JT, Yang J, Chen DC, Zhang XL, Tang ZS. An optimized mini-preparation method to obtain high-quality genomic DNA from mature leaves of sunflower. Genet Mol Res. 2007;6:1064–71.
21. Weising K, Nybom H, Pfenninger M, Wolff K, Meyer W. DNA fingerprinting in plants and fungi. Boca Raton (FL): CRC Press; 1994.
22. Arumuganathan K, Earle ED. Nuclear DNA content of some important plant species. Plant Mol Biol Rep. 1991;9:208–18. [CrossRef]
23. Cheng X, Hong X, Khayatnezhad M, Ullah F. Genetic diversity and comparative study of genomic DNA extraction protocols in Tamarix L. species. Caryologia. 2021;74:131–9. [CrossRef]
24. Dairawan M, Shetty PJ. The evolution of DNA extraction methods. Am J Biomed Sci Res. 2020;8:39–46. [CrossRef]
25. Michiels AN, Van den Ende W, Tucker M, Van Riet L, Van Laere A. Extraction of high-quality genomic DNA from latex-containing plants. Anal Biochem. 2003;315:85–9. [CrossRef]
26. Heikrujam J, Kishor R, Mazumder PB. The chemistry behind plant DNA isolation protocols. Biochem Anal Tools–Methods Bio-Molecules Stud. 2020;8:131–41.
27. Abhijit S, Manjushree D. Standardization of DNA extraction and optimization of RAPD-PCR conditions in Garcinia indica. Int J Bot. 2010;6;293–8. [CrossRef]
28. Shah A, Gadol N, Priya G, Mishra P, Rao M, Singh NK, et al. Morpho-physiological and metabolites alteration in the susceptible and tolerant genotypes of sesame under waterlogging stress and post-waterlogging recovery. Plant Stress. 2024;11:100361.
