
1. INTRODUCTION
Iron is an essential element involved in various biochemical and metabolic processes, including oxygen transport, energy transduction, and DNA synthesis [1]. However, like many other micronutrients, high doses of iron are associated with harmful side effects. Iron overload refers to the accumulation of iron in organs, particularly the liver, pancreas, and heart, that causes various organ failures [2,3] and several disorders, including beta-thalassemia [4,5], type 2 diabetes mellitus (T2DM), and nonalcoholic fatty liver disease (NAFLD) [6]. A recent study reported that the major contributors to mortality in patients with iron overload are infection and cardiac complications [7]. Under normal conditions, iron is bound to transferrin, and the body stores it in the form of ferritin to maintain physiological levels of iron in the body [8]. During iron overload, the capacity of transferrin to bind iron is exceeded, leading to the subsequent formation of non-transferrin-bound iron (NTBI), which is taken up non-selectively by cells via NTBI transporters on the cell membrane [9]. The formation of NTBI increases the cellular labile iron pool (LIP) and catalyzes the formation of reactive oxygen species (ROS), including the superoxide anion (O2•), hydrogen peroxide (H2O2), and hydroxyl radical (HO•), through the Fenton and Haber–Weiss reactions, which initiate oxidative stress [10,11]. The excessive generation of ROS causes oxidative stress, which induces molecular damage and impairs cellular functions [12,13]. Therefore, effective new therapies have been developed to target and prevent iron overload and toxicity.
Chelation therapy has long been used as a treatment for diseases related to metal ion poisoning, either as primary, alternative, or adjuvant therapy [14]. Iron-chelating agents such as deferoxamine (DFO), deferiprone, and deferasirox have been introduced in medical care to protect patients from iron toxicity [15]. These chelating drugs are used daily to ameliorate iron overload in thalassemia [16]. DFO, the gold standard in iron chelation therapy over the past 50 years [17], specifically binds to six ligands of Fe3+, rendering it stable against reactions with free radicals [18], which then attenuates iron-induced oxidative stress by binding to free iron and preventing it from catalyzing the formation of ROS [19,20]. A recent report has demonstrated that DFO ameliorates iron overload and reduces ROS in a traumatic brain injury-induced rat model [21]. In clinical settings, DFO is given parenterally due to its short plasma half-life [22]. Most of the clinically used chelating agents, including DFO, have adverse side effects and a narrow scope of applications [23]. A recent study showed that individuals who are susceptible to transfusion-related iron overload may develop idiosyncratic drug-induced neutropenia after treatment with chelating agents, which could increase their risk of infection [24]. Additionally, DFO causes ocular and auditory neurotoxicity, pulmonary and renal toxicity, gastrointestinal problems, muscle spasms and pain, and growth retardation [15]. Hence, new treatment modalities are to be discovered, for which many classes of phytochemical-based chelators with additional antioxidant activities hold great promise [25,26].
Sulforaphane (SFN) is an aliphatic isothiocyanate that is mainly found in cruciferous vegetables such as broccoli and Brussels sprouts. Recent studies have reported that SFN protects human mesenchymal stem cells and mononuclear cells from cadmium toxicity [27,28]. SFN exerts cytoprotective effects by reinforcing the transcription factor called nuclear factor erythroid 2-related factor 2 (Nrf2) [29,30]. In fact, SFN is known as one of the most potent and rapid inducers of Nrf2 [31,32]. Nrf2 serves as a central regulator of the antioxidant response network against oxidative stress through the regulation of antioxidant response element (ARE)-mediated gene induction, including heme oxygenase 1 (HO-1) and NAD(P)H quinone dehydrogenase 1 (NQO1) [33-35]. Additionally, recent findings suggest that Nrf2 induces iron-metabolizing enzymes [1,36,37]. Collectively, those reports prompted us to hypothesize that SFN protects cells from iron toxicity by activating the Nrf2 system.
In this study, we used FeCl2 (Fe2+) and FeCl3 (Fe3+) to trigger iron overload in vitro, examined the effects of SFN on intracellular ROS levels and cell viability, and elucidated its mechanism of action. This study provided a novel strategy for reducing oxidative stress under excess iron conditions.
2. METHODS
2.1. Cell Cultures and Treatments
The human hepatoma cell line (Hep3B) was cultured in DMEM (Wako, Osaka, Japan) supplemented with FBS, penicillin, and streptomycin and incubated at 37°C in 95% air and 5% CO2. Hep3B cells were treated with iron (II) chloride tetrahydrate (FeCl2.4H2O; Wako) and iron (III) chloride hexahydrate (FeCl3.6H2O; Wako) for 36 h at a final concentration of 200 μM, which is an equivalent concentration of iron ions in human livers with thalassemia [38]. DFO (Wako) at a concentration of 200 µM, SFN (Cayman Chemical Company, Ann Arbor, MI) at a concentration of 10 or 25 µM, and a PI3K inhibitor (LY294002; Wako) at a concentration of 50 µM were added to Hep3B cells for 8, 30, and 30 h, respectively.
2.2. Preparation of Construct and Transfection
Human Nrf2 (NM_006164) cDNA was amplified by polymerase chain reaction (PCR) using primers 5′AAGGATCCATCATGATGGACTTGGAGCT-3′ (forward; underlined, BamHI site) and 5′TTTCTAGACTAGTTTTTCTTAACATC-3′ (reverse; underlined, XbaI site). Human MT (NM_005953) cDNA was amplified by PCR using primers 5′AAGAATTCTGATCCCAACTGCTCCTG-3′ (forward; underlined, EcoRI site) and 5′TTTCTCGAGTCAGGCGCAGCAGCTGCACT-3′ (reverse; underlined, XhoI site). Amplified Nrf2 and MT were then digested with BamHI and XbaI, and EcoRI and XhoI, respectively, and inserted into the 3×FLAG-pcDNA4 and pcDNA3.1(+) vectors (Invitrogen, Carlsbad, CA). Nrf2- and MT-containing plasmids were transfected into cells using standard calcium phosphate. For the knockdown experiments, si-Nrf2, si-MT, and AllStars Negative Control were purchased from Qiagen (Hilden, Germany) and were transfected with ScreenFect™ A (Wako, Osaka, Japan) according to the manufacturer’s instructions.
2.3. ROS Measurement
The intracellular level of ROS was measured by 2′7′dichlorodihydrofluorescein diacetate (DCFH-DA; Sigma Aldrich Co., St. Louis, MO) according to the previously reported method [39]. Hep3B cells were incubated in a medium containing 10 nM DCFH-DA, an intracellular probe for oxidative stress, for 10 min in the dark at 37°C [40]. Cells were washed with PBS, and fluorescence intensity was measured using a plate reader (EnVision 2104 Multilabel Reader, PerkinElmer, Waltham, MA) with an excitation wavelength of 480 nm and an emission wavelength of 530 nm.
2.4. MTT Assay
MTT assay was used for the measurement of cell viability according to the previously reported methods [41]. In brief, around 2 × 104 cells were incubated in the 12-well plate dish. After corresponding treatment, 5 mg/mL MTT (3-(4,5-di-methylthiazol-2-yl)-2,5-diphenyltetrazolium bromide; Sigma) was added to the medium, and cells were incubated for 2 h at 37oC. The culture medium was removed, and the resulting purple formazan was dissolved in 500 μL of isopropanol containing 0.04N HCl and 0.1% NP40. The absorbance was measured at 590 nm using a microplate reader (PerkinElmer, Waltham, MA).
2.5. Immunofluorescence
Hep3B cells were cultured in 3.5-cm glass-bottomed dishes (Thermo Fisher Scientific, Waltham, MA), treated with corresponding treatment, washed with PBS, and fixed with 4% paraformaldehyde (Wako) at 4°C for 20 min, followed by washing with PBS containing 0.2% Tween 20 (TPBS) (Bio-Rad, Hercules, CA). Blocking was performed using 0.1% bovine serum albumin (Wako) diluted in TPBS at 4°C for 1 h. Cells were then incubated with the anti-Nrf2 antibody (1: 1,000) at 4°C for 1 h and washed three times with TPBS. Cells were incubated with Alexa Fluor® 488-conjugated goat anti-rabbit IgG (1: 1,000; Invitrogen) at 4°C for 1 h, then washed three times with TPBS. The nucleus was counterstained using 4′,6-diamidino-2-phenylindole (DAPI; 1:1,000; Dojindo, Kumamoto, Japan). Immunofluorescence was detected using the confocal microscope TCS SP8 (Leica Microsystems, Wetzlar, Germany).
2.6. Western Blotting
Whole-cell homogenates were subjected to Western blotting using antibodies against Nrf2 and β-actin. The antibodies against Nrf2 and β-actin were prepared as previously described [42]. Proteins were loaded and separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) on 7.5% SDS polyacrylamide gel and then transferred to nitrocellulose membranes (Bio-Rad Laboratories, Hercules, CA). Membranes were incubated with the primary antibodies, and proteins were visualized by horseradish peroxidase conjugated to goat anti-rabbit or anti-mouse IgG (secondary antibody) and 4-chloro-1-naphthol (4CN; Bio-Rad Laboratories). The band intensities of proteins were quantified using ImageJ (Version 1.36b; National Institutes of Health, Bethesda, MD).
2.7. Isolation of RNA and Reverse Transcription PCR
Total RNA was extracted from Hep3B cells using Isogen in accordance with the manufacturer’s instructions. Total RNA was converted to cDNA by MMLV Reverse Transcriptase (Nippon Gene, Toyama, Japan) with the following protocol: incubation at 25°C for 15 min, 42°C for 60 min, followed by heating at 70°C for 10 min. PCR was performed using Go Taq Green Master Mix (Promega, WI), 10 pmol of each primer, and 100 ng of cDNA at 94°C for 30 s, 55°C for 30 s, and 72°C for 30 s. All primers and GenBank accession numbers are shown in Table 1. PCR products were separated by electrophoresis on a 1% agarose gel and visualized by ethidium bromide staining. The band intensities were quantified using ImageJ (Version 1.36b; National Institutes of Health, Bethesda, MD).
Table 1: Primers used for the gene expression analysis.
| Primers | GenBank Accession No. | Sequences | |
|---|---|---|---|
| HO-1 | NM_002133 | Forward | 5′- CCAGCCATGCAGCACTATGT -3′ |
| Reverse | 5′- AGCCCTACAGCAACTGTCGC -3′ | ||
| NQO1 | NM_000903 | Forward | 5′- TGATCGTACTGGCTCACTCA -3′ |
| Reverse | 5′- GTCAGTTGAGGTTCTAAGAC -3′ | ||
| FTH | NM_002032 | Forward | 5′-ATCTTCCTTCAGGATATCAA-3′ |
| Reverse | 5′-ATCACTGTCTCCCAGGGTGT-3′ | ||
| MT | NM_005953 | Forward | 5′-ATGGATCCCAACTGCTCCTG-3′ |
| Reverse | 5′-TCAGGCGCAGCAGCTGCACT-3′ | ||
| β-actin | NM_001101 | Forward | 5′- CAAGAGATGGCCACGGCTGCT -3′ |
| Reverse | 5′- TCCTTCTGCATCCTGTCGGCA -3′ |
2.8. Bioinformatics Analysis on Differentially Expressed Genes
The transcriptomic datasets were screened from the publicly available Gene Expression Omnibus (GEO, http://www.ncbi.nlm.nih.gov/geo) [43] database with the keywords “iron overload,” “Nrf2,” “sulforaphane,” “mRNA,” “liver,” and “Homo sapiens.” The GSE186655, GSE230608, and GSE20479 were selected. The raw data were processed using a built-in interactive online tool, GEO2R (https://www.ncbi.nlm.nih.gov/geo/geo2r/) [43]. Differentially expressed genes (DEGs) were screened with a false discovery rate (FDR) corrected p-value <0.05, and |fold change (FC)| >2.0 was selected as the screening threshold for each group. The DEGs in each dataset were presented in volcano plots and integrated into Venn diagrams using an online platform for data analysis and visualization SRplot (http://www.bioinformatics.com.cn/srplot).
2.9. Statistical Analysis
All values are given as the mean ± S.D. and analyzed with IBM SPSS Statistics for Windows, version 23.0 (IBM Corp., Armonk, NY, USA). p-values were assessed by the Student’s t-test or a one-way repeated measures ANOVA, followed by respective post-hoc tests for multiple comparisons against specified groups as described in the figure legends. Differences were considered significant when p was <0.05.
3. RESULTS
3.1. Iron Overload Induces Oxidative Stress in Hep3B Cells
We found that FeCl2 (Fe2+) and FeCl3 (Fe3+) at a concentration of 200 μM for 36 h treatments in Hep3B cells increased intracellular ROS levels [Figure 1A]. The elevation of ROS may induce lipid peroxidation, DNA damage, and protein modifications, which decrease cell viability and ultimately result in cell death [44]. Therefore, we also examined the viability of cells using the MTT assay, and we found that Fe2+ and Fe3+ treatments decreased the viability of Hep3B cells [Figure 1B]. Since the intracellular response to high ROS levels is orchestrated by Nrf2, we measured the protein levels of Nrf2 after the Fe2+ and Fe3+ treatments. The results showed that Fe2+ and Fe3+ induced approximately a threefold and sevenfold increase in the accumulation of the Nrf2 protein, respectively [Figure 1C]. Increases in Nrf2 levels are generally accompanied by the nuclear localization of Nrf2 [45,46]. Indeed, Fe2+ and Fe3+ treatments promoted the nuclear localization of Nrf2 [Figures 1D–F]. Consistently, the activity of Nrf2 was enhanced by Fe2+ and Fe3+ treatments, as indicated by the elevated expression of its putative target genes (HO-1 and NQO1) [Figure 1G]. Regarding iron metabolism, a recent study suggested that Nrf2 also regulates the expression of the ferritin heavy chain (FTH), which plays a role in the storage of iron [35]. We also found that excess iron treatment significantly increased FTH mRNA [Figure 1G].
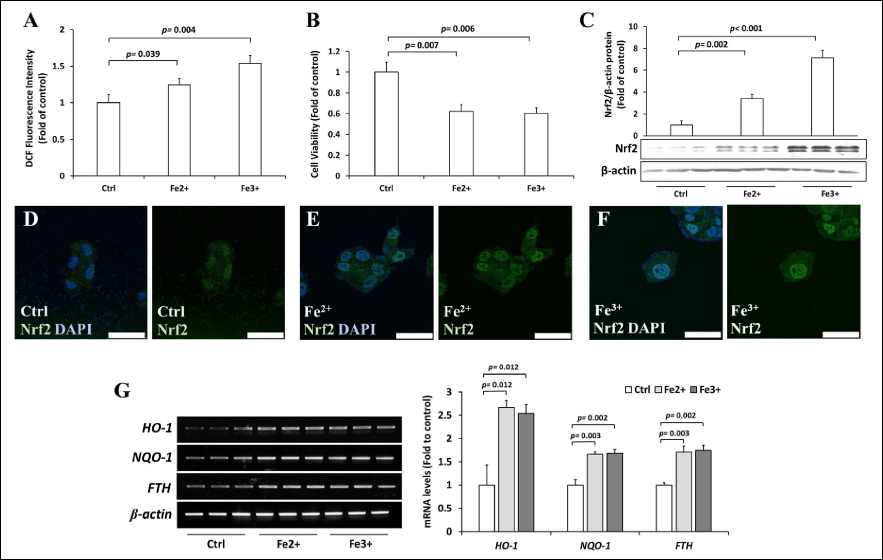 | Figure 1: Effects of Fe2+ and Fe3+ ions on Hep3B cells. (A) Hep3B cells were treated with 200 μM Fe2+ and Fe3+ for 36 h, and the intracellular ROS levels were measured by DCFH-DA assay. (B) Hep3B cells were treated with 200 μM Fe2+ and Fe3+ for 36 h, and the viability of cells was examined using the MTT assay. (C) Hep3B cells were treated with 200 μM Fe2+ and Fe3+ for 36 h, and the expression of Nrf2 was assessed by Western blotting. (D–F) Hep3B cells were treated with vehicle (D), Fe2+ (200 μM, 36 h) (E), and Fe3+ (200 μM, 36 h) (F), and the cellular localization of Nrf2 (green) was observed by immunofluorescence counterstained with DAPI (blue). (G) Hep3B cells were treated with 200 μM Fe2+ and Fe3+ for 36 h, and the expressions of HO-1, NQO1, and FTH mRNAs were measured by RT-PCR. Values are mean ± S.D. relative to the corresponding treatments with vehicle (Ctrl, designated as 1). Differences were analyzed by one-way repeated measures ANOVA with Dunnett’s post-hoc test (n = 3). [Click here to view] |
3.2. DFO Does Not Improve the Cell Viability of Iron-treated Hep3B Cells
DFO is an FDA-approved, gold-standard therapy for acute iron intoxication and chronic iron overload due to transfusion-dependent anemia or thalassemia. To support its clinical efficacy, we examined the effects of DFO on oxidative stress and the cell viability of excess iron-induced Hep3B cells. We observed that DFO treatment significantly decreased intracellular ROS levels in Fe3+-treated cells but not in Fe2+-treated cells [Figure 2A], and DFO treatment did not affect the viability of cells [Figure 2B]. Next, we examined the effects of DFO on the activity of the Nrf2 pathway. DFO significantly reduced Nrf2 protein [Figures 2C and 2D] and the expression of Nrf2 target genes [Figure 2E] in Fe2+- and Fe3+-treated cells. These results suggested that the iron chelator DFO can be useful in preventing oxidative stress in iron overload conditions, but for unknown reasons, DFO does not recover the viability of cells.
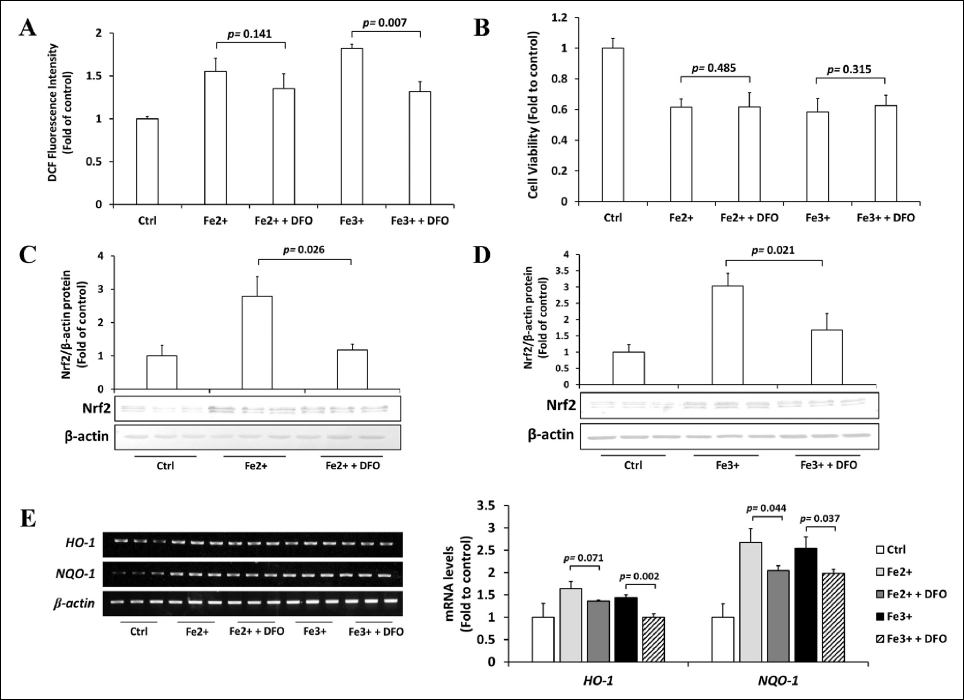 | Figure 2: Effects of DFO on excess iron-treated Hep3B cells. (A) Hep3B cells were treated with 200 μM DFO for 8 h, followed by 36 h incubation with 200 μM Fe2+ or Fe3+, and the intracellular ROS levels were measured by DCFH-DA assay. (B) Hep3B cells were treated with 200 μM DFO for 8 h, followed by 36 h incubation with 200 μM Fe2+ or Fe3+, and the viability of cells was examined using the MTT assay. (C & D) Hep3B cells were treated with 200 μM DFO for 8 h, followed by 36 h incubation with 200 μM Fe2+ (C) or Fe3+ (D), and the expression of Nrf2 was assessed by Western blotting. (E) Hep3B cells were treated with 200 μM DFO for 8 h, followed by 36 h incubation with 200 μM Fe2+ or Fe3+, and the expressions of HO-1 and NQO1 mRNAs were measured by RT-PCR. Values are mean ± S.D. relative to the corresponding treatments with vehicle (Ctrl, designated as 1). Differences were analyzed by one-way repeated measures ANOVA with Tukey’s post-hoc test (n = 3). [Click here to view] |
3.3. SFN Mitigates Iron Toxicity in Hep3B Cells by Upregulating Nrf2
To gain insight into the role of nutraceuticals present in vegetables on iron toxicity, we selected SFN due to its potential to alleviate oxidative stress. We first confirmed the long-standing theory that SFN activates Nrf2 levels. As expected, SFN induced Nrf2 protein levels [Figure 3A], induced Nrf2 nuclear accumulation [Figure 3B], and activated the expression of HO-1 and NQO-1 [Figure 3C]. Next, we examined whether SFN protects Hep3B cells from iron toxicity. We found that SFN significantly reduced the intracellular ROS [Figure 3D] and improved cell viability [Figure 3E] of excess iron-induced Hep3B cells. Several studies have shown that the Nrf2-activating effects of SFN are mediated by phosphoinositide 3-kinase (PI3K) [47,48]. Indeed, the PI3K inhibitor, LY294002, partially inhibited the activation of Nrf2 by SFN [Figure 3F].
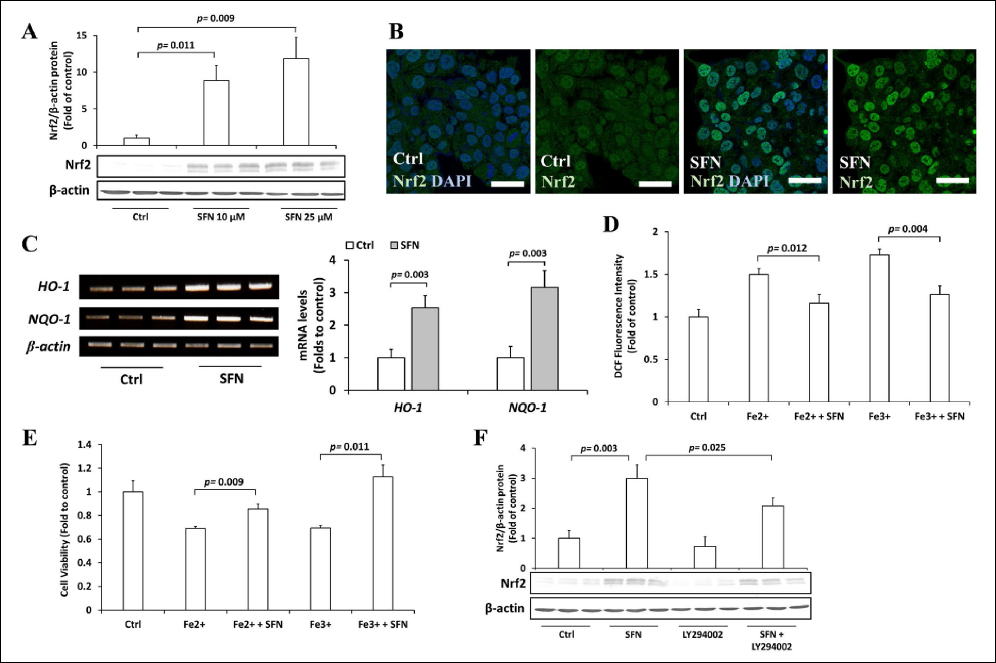 | Figure 3: Role of SFN on excess iron-treated Hep3B cells. (A) Hep3B cells were treated with 10 or 25 μM SFN for 30 h, followed by 36 h incubation with 200 μM Fe2+ or Fe3+, and the expression of Nrf2 was assessed by Western blotting. (B) Hep3B cells were treated with 10 μM SFN for 30 h, and the cellular localization of Nrf2 (green) was observed by immunofluorescence counterstained with DAPI (blue). (C) Hep3B cells were treated with 10 μM SFN for 30 h, and the expressions of HO-1 and NQO1 mRNAs were measured by RT-PCR. (D) Hep3B cells were treated with 10 μM SFN for 30 h, followed by 36 h incubation with 200 μM Fe2+ or Fe3+, and the intracellular ROS levels were measured by DCFH-DA assay. (E) Hep3B cells were treated with 10 μM SFN for 30 h, followed by 36 h incubation with 200 μM Fe2+ or Fe3+, and the viability of cells was examined using the MTT assay. (F) Hep3B cells were treated with 10 μM SFN, 50 μM LY294002, or both for 30 h, and the expression of Nrf2 was assessed by Western blotting. Values are mean ± S.D. (n = 3) relative to the corresponding treatments with vehicle (Ctrl, designated as 1). Differences to this value were analyzed by one-sample t-test with Bonferroni’s correction for multiple testing (for C) and one-way repeated measures ANOVA with Dunnett’s (for A) or Tukey’s (for D–F) post-hoc tests. [Click here to view] |
Next, we explored whether SFN-regulated Nrf2 protein abundance is the critical mechanism for its protective activity against iron-induced toxicity. To explore this, we first observed the effects of SFN on the Nrf2 pathway under the excess iron condition. SFN further increased the abundance of Nrf2 protein in Fe2+- and Fe3+-treated Hep3B cells [Figures 4A and 4B]. The expression of Nrf2-target genes HO-1 [Figure 4C] and NQO-1 [Figure 4D] mRNAs was further elevated by SFN in Fe2+- and Fe3+-treated cells. Second, we observed that Nrf2 knockdown by siRNA completely diminished the effects of SFN on intracellular ROS levels [Figure 4E] and cell viability [Figure 4F]. Together, we unequivocally proved the critical involvement of the Nrf2 pathway and that SFN protects cells from iron toxicity by further activating Nrf2.
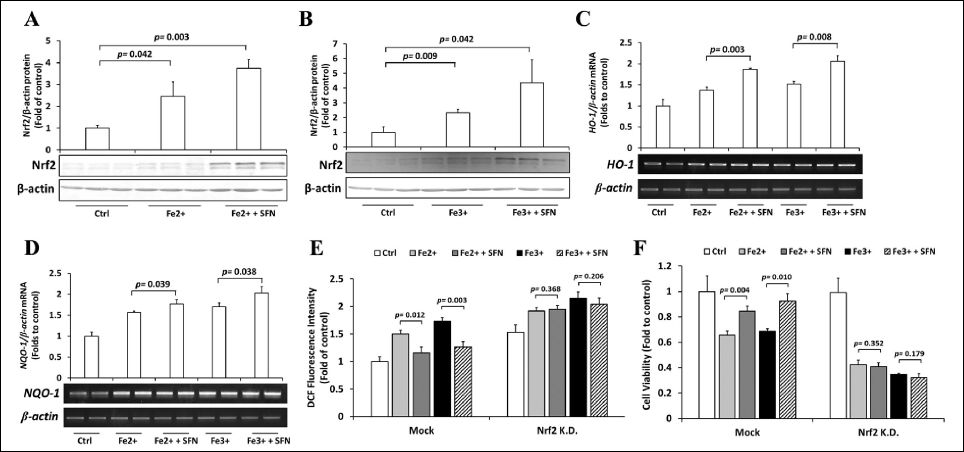 | Figure 4: Role of Nrf2 on cytoprotective effects of SFN. (A & B) Hep3B cells were treated with 10 μM SFN for 30 h, followed by 36 h incubation with 200 μM Fe2+ (A) or Fe3+ (B), and the expression of Nrf2 was assessed by Western blotting. (C & D) Hep3B cells were treated with 10 μM SFN for 30 h, followed by 36 h incubation with 200 μM Fe2+ or Fe3+, and the expressions of HO-1 (C) and NQO1 (D) mRNAs were measured by RT-PCR. (E & F) Hep3B cells were transfected with siRNA-control or siRNA-Nrf2, treated with 10 μM SFN for 30 h, followed by 36 h incubation with 200 μM Fe2+ or Fe3+. The intracellular ROS levels (E) and the viability of cells (F) were examined using the DCFH-DA and MTT assays, respectively. Values are mean ± S.D. relative to the corresponding treatments with vehicle (Ctrl, designated as 1). Differences to this value were analyzed by one-way repeated measures ANOVA with Dunnett’s (for A & B) or Tukey’s (for C–F) post-hoc tests. [Click here to view] |
3.4. SFN Activates Nrf2-Regulated MT to Protect Hep3B Cells From Iron Toxicity
To gain a deeper insight into the mechanism by which SFN and Nrf2 protect cells from iron toxicity, we explored publicly available gene expression datasets by using the GEO [http://www.ncbi.nlm.nih.gov/geo] database and selected datasets with the accession numbers GSE186655, GSE230608, and GSE20479. The GSE186655 contains RNA-seq data from three control and three ferric ammonium citrate-treated HepG2 cells [49]. The GSE230608 contains RNA-seq data of control (n = 3) and Keap1-knockout (n = 3) HepG2 cells [50]. The GSE20479 contains microarray data of control (n = 4) and SFN-treated (n = 4) primary human hepatocytes [51]. Following the screening of data based on the FDR-corrected p-value <0.05 and |FC| >2.0, the number of DEGs obtained was 566 in GSE186655, 560 in GSE230608, and 1226 in GSE20479. In GSE186655, we observed 344 upregulated and 222 downregulated genes [Figure 5A]. In GSE230608, we found 404 upregulated and 156 downregulated genes [Figure 5B]. In GSE20479, we found 525 upregulated and 701 downregulated genes [Figure 5C]. The Venn diagram was used to identify overlapping genes in all sets. Nine genes were identified as common DEGs in iron overload conditions, Nrf2 overexpression, and SFN treatment. We identified metallothionein (MT), a metal-binding protein, as a commonly upregulated gene [Figure 5D].
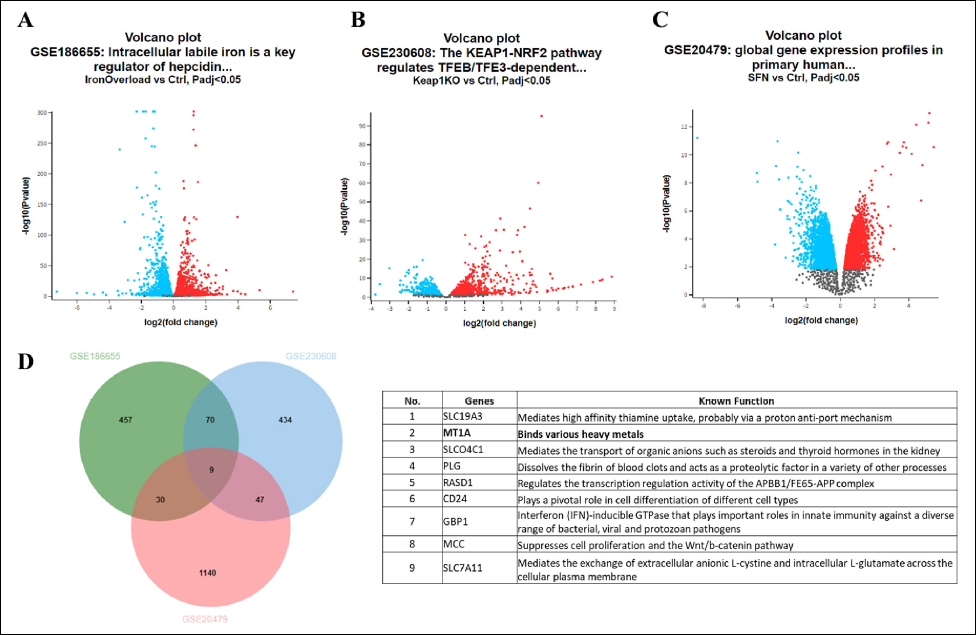 | Figure 5: Differentially expressed genes (DEGs) in iron overload, Nrf2 overexpression, and SFN treatment datasets. (A–C) Volcano plot of DEGs in GSE186655 (A), GSE230608 (B), and GSE20479 (C) datasets. Upregulated and downregulated genes are shown in red and blue, respectively. (D) Overlapping DEGs among three GEO datasets represented through a Venn diagram (left panel). The physiological functions of the DEGs are obtained from GeneCards (https://www.genecards.org/) and presented (right panel). [Click here to view] |
Next, we examined the expression of MT mRNA under excess iron, Nrf2 overexpression, and SFN treatment to experimentally prove the involvement of MT. Excess iron (both Fe2+ and Fe3+) increased the expression of MT mRNA by approximately 50% [Figure 6A]. The overexpression of Nrf2 induced a twofold increase in the accumulation of MT mRNA [Figure 6B]. Finally, SFN elevated the abundance of MT mRNA by approximately 40% [Figure 6C]. To assess the function of MT against iron toxicity, we constructed and transfected FLAG-tagged MT into iron-treated Hep3B cells. We observed that MT overexpression reduced intracellular ROS levels in excess iron-treated cells [Figure 6D] and was sufficient to restore the viability of cells [Figure 6E]. In conclusion, we showed that SFN, via Nrf2, activates MT transcriptionally to reduce excess iron toxicity.
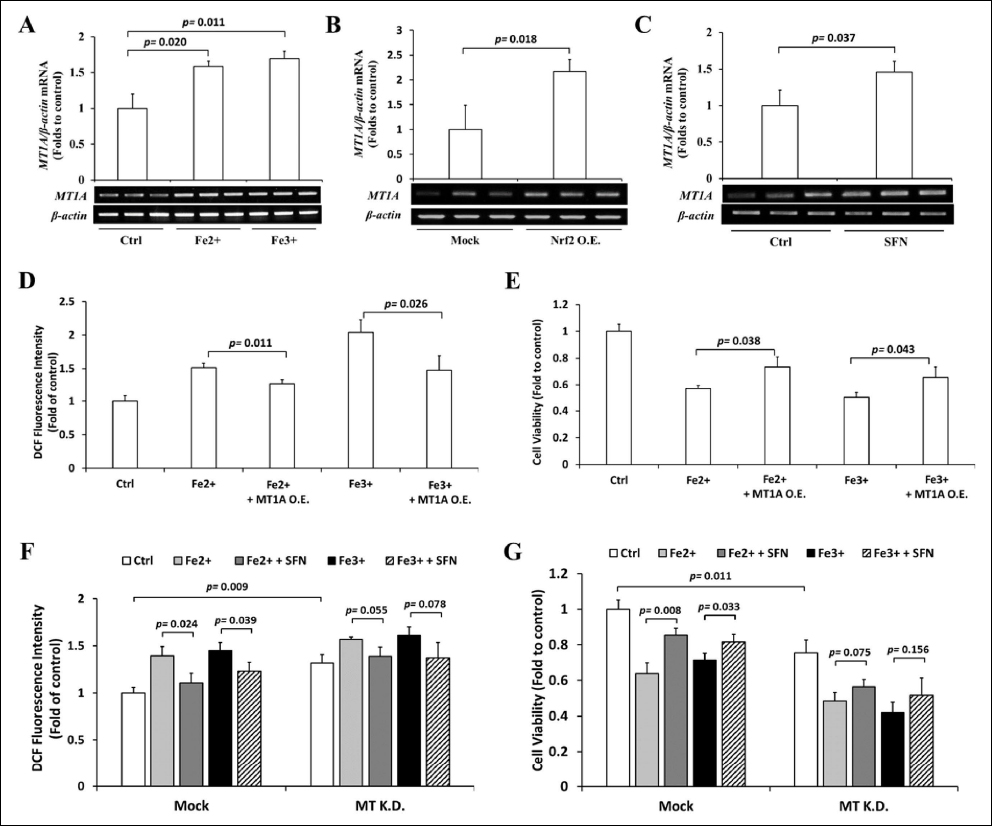 | Figure 6: Role of MT on excess iron-treated Hep3B cells. (A) Hep3B cells were treated with 200 μM Fe2+ or Fe3+ for 36 h, and the expression of MT mRNA was assessed by RT-PCR. (B) Nrf2 in pcDNA3.1(+) was transfected in Hep3B cells, and the mRNA level of MT was evaluated by RT-PCR. (C) Hep3B cells were treated with 10 μM SFN for 30 h, and the expression of MT mRNA was measured by RT-PCR. (D & E) Hep3B cells transfected with FLAG-mock or FLAG-MT were treated with 10 μM SFN for 30 h, followed by 36 h incubation with 200 μM Fe2+ or Fe3+, and the intracellular ROS levels (D) and the viability of cells (E) were examined using the DCFH-DA and MTT assays, respectively. (F & G) Hep3B cells were transfected with siRNA-control or siRNA-MT, treated with 10 μM SFN for 30 h, followed by 36 h incubation with 200 μM Fe2+ or Fe3+. The intracellular ROS levels (F) and the viability of cells (G) were examined using the DCFH-DA and MTT assays, respectively. Values are mean ± S.D. relative to the corresponding treatments with vehicle (Ctrl, designated as 1). Differences to this value were analyzed by one-sample t-test (for B & C) and one-way repeated measures ANOVA with Dunnett’s (for A) or Tukey’s (for D–G) post-hoc tests. [Click here to view] |
As iron overload and Nrf2 can both induce MT expression, the cellular responses are completely opposite. While iron overload leads to cell death, Nrf2 activation can be protective against iron overload-induced cell death. We hypothesize that an increase in MT expression may not be a key event in iron overload-induced cell death. To clarify this phenomenon, we further explore the role of MT activation in the cellular response to iron overload. We found that MT knockdown by siRNA elevated basal ROS levels and partially exacerbated iron overload-induced oxidative stress and cell death, suggesting that the activation of MT is an important cytoprotective mechanism against iron overload. Next, we added SFN to iron overload-treated MT knockdown cells and showed that the protective roles of SFN on oxidative stress [Figure 6F] and cell viability [Figure 6G] were partially attenuated. Together, these findings suggest that Nrf2-mediated MT expression is a critical event to prevent iron overload-induced cell death. The activation of the cellular Nrf2/MT axis solely by iron overload may not be sufficient to protect cells; therefore, further activation of the Nrf2/MT axis by an additive such as SFN provides more protection against cell damage.
4. DISCUSSION
Iron is an essential mineral that participates in a wide variety of metabolic processes. Humans ingest two types of iron through their diet: heme and non-heme. Meat, poultry, and fish contain heme, whereas plant foods such as leafy greens, legumes, nuts, and seed grains contain non-heme iron. Although essential, iron can also be toxic in excess and can be life-threatening [52]. Hereditary hemochromatosis, blood transfusion, hemolysis, and excessive parenteral or dietary consumption can cause iron overload [53]. Iron overload causes excessive iron deposition in a wide variety of tissues because humans do not have the ability to actively excrete iron. For instance, in beta-thalassemia, excess iron resulting from chronic blood transfusions containing 200–250 mg of iron/unit-packed red blood cells is deposited in organ tissues, particularly the liver, which leads to oxidative damage [38,54,55]. In the present study, we showed that iron overload leads to oxidative damage in the hepatoma cell line, Hep3B. Elevated cellular LIP during iron overload catalyzes the formation of ROS through the Fenton and Haber–Weiss reactions [10,11]. Various chelating compounds, including DFO, DFP, and DFX, have been used in clinical settings to inhibit iron uptake, improve iron excretion, and reduce iron toxicity [15]; nevertheless, chelation therapy is not very effective or safe [23]. In fact, we found that DFO did not improve the viability of Hep3B cells under excess iron conditions. Therefore, exploring a diet-based treatment is required to prevent and reduce the effects of iron overload toxicity.
In response to elevated radicals, the transcription factor Nrf2 is activated, acting as the converging point of these stimuli [46,56]. Exposure to electrophiles and oxidants attenuates the activity of Kelch-like ECH-associated protein 1 (Keap1), which is a major regulator of Nrf2. Keap1 acts as an adaptor of the Cullin 3-based E3 ubiquitin ligase that targets Nrf2 for posttranslational modification in the form of ubiquitination, causing subsequent proteasomal degradation of Nrf2. Electrophiles and oxidants modify the structure of Keap1, leading to the inhibition of Nrf2 degradation and subsequently promoting Nrf2 translocation into the nucleus, where it activates the transcription of cytoprotective genes against environmental stresses [57-59]. Regarding iron metabolism, Nrf2 transcriptionally activates the iron storage proteins ferritin (FTH) and heme oxygenase 1 (HO-1), which leads to a net deregulation of iron metabolism [60]. Previous studies reported that Nrf2 knockout mice are more vulnerable to iron-induced liver oxidative injury [61].
Due to the importance of Nrf2 in various diseases [62], many studies have focused on exploring potent Nrf2 modulators to combat oxidative damage, inflammation, and metabolic abnormalities. Currently, there are approximately 100 clinical trials with Nrf2 activators listed on Clinicaltrials.gov in the form of pure substances, dietary supplements, or plant extracts. The naturally occurring isothiocyanate SFN, the semisynthetic cyanoenone triterpenoid RTA-408 (omaveloxolone), and dimethyl fumarate (DMF, Tecfidera®) are the only Nrf2 activators currently used in clinical practice [63]. In the present study, we looked at how SFN affected the iron-induced toxicity in Hep3B cells. Cruciferous vegetables are rich in SFN, a chemical with anti-inflammatory, anti-cancer, anti-microbial, and antioxidant properties [64]. SFN is one of the most powerful and rapid activators of Nrf2 [65]. Multiple studies have shown that SFN exerts protective effects against oxidative stress-induced cellular injury by inducing Nrf2 and the expression of its downstream genes [31]. Therefore, we postulated that SFN may reduce iron-induced oxidative toxicity patterns in the human liver.
The effectivity of SFN was examined by adding SFN to iron-treated cells to investigate its protective roles. The results revealed that SFN treatment decreased intracellular ROS levels, and, intriguingly, SFN also improved the viability of excess iron-treated Hep3B cells. These findings unequivocally demonstrate that SFN is a biocompatible substance that is excellent for use in the clinical context of iron overload-related conditions. These data are supported by previous findings showing that SFN significantly alleviated oxidative stress-induced damage through the PI3K-Akt-mediated activation of Nrf2 [66]. In the present study, we have also demonstrated that Nrf2 knockdown completely diminished the effects of SFN on the elevated intracellular ROS levels and reduced cell viability caused by iron overload. Taken together, these results imply that SFN plays a cytoprotective role in attenuating Fe ion-induced oxidative stress by activating Nrf2 through pathway(s) other than the canonical ROS-mediated activation of Nrf2 involving Keap1 [67]. Various studies have indicated that Nrf2 can control the storage and outflow of iron through the regulation of transcription of ferritin (an iron storage protein) and ferroportin 1 (an iron exporter protein) [1]. Additionally, the regulation of HO-1 gene expression by Nrf2 is critical for the cellular response to iron overload. Furthermore, through the actions of bilirubin, HO-1 demonstrates anti-oxidative functions [68]. Numerous recent studies indicate that the cytoprotective effects against ferroptosis are related to activation of the Nrf2/HO-1 axis [69-71], which is consistent with this study.
We next examined the publicly available transcriptomic datasets to gain mechanistic insight into the effects of SFN and Nrf2 on iron metabolism. We found that excess iron, Nrf2, and SFN upregulated MT. MT is a thiol-rich and heavy metal-binding protein that contains 20 cysteine residues and binds to metal ions through its thiol (-SH) group. It acts as an antioxidant by scavenging free radicals, enhancing cytoprotection against oxidative stress [72-75]. MT has been documented to bind heavy metals such as copper (Cu) with the greatest stability constant, followed by cadmium (Cd), zinc (Zn), mercury (Hg), and bismuth (Bi) [76]. The induction of MT is generally associated with heavy metals, such as zinc (Zn) ions [77]; however, limited information is currently available on the relationship between MT and Fe ions.
The present study clearly demonstrated that the overexpression of MT was sufficient to reduce the generation of intracellular ROS and improve cell viability, suggesting that MT has the capability to ameliorate iron overload-related pathology. Our findings are supported by Baird and colleagues, who reported that upregulated MT protects murine macrophage-like J774 cells from apoptosis following oxidative stress by stabilizing lysosome function [20]. In iron-induced dopaminergic neurons, MT prevents oxidative stress by interacting with glutathione (GPx), while oxidized glutathione regulates zinc transfer from MT to zinc-depleted enzymes. Therefore, MT and GPx complement each other’s functions during iron-induced oxidative stress [78]. A study in rats’ blood cells suggested a reciprocal association between dietary iron consumption and MT expression, even though MT does not directly mobilize iron. This relationship may be indicative of alterations in cellular Zn homeostasis or redox status. An MT-Zn axis may very well be functional in sizing the Zn pool, in addition to processes that control iron homeostasis, as excess iron interferes with Zn availability to proteins and vice versa [79].
Studies have reported that MT plays a prominent role in mediating the protective role of SFN against renal and cardiac injury caused by oxidative stress via the upregulation of Nrf2 [80,81]. Additionally, the study suggested that the activation of Nrf2-driven MT gene expression by SFN is mediated by mitogen-activated protein kinase (MAPK) in human hepatoma HepG2 cells, which is related to the anti-cancer activity of SFN [82]. In the present study, we showed the involvement of PI3K, instead of MAPK, in the regulation of Nrf2 protein abundance by SFN. Taken together, the concurrent activation of Nrf2 and elevation of MT following the induction of SFN serve as a protective mechanism against oxidative damage in iron overload conditions.
In conclusion, we herein demonstrate that excess iron-induced oxidative toxicity can be attenuated by SFN, which induces Nrf2 and subsequently increases MT at transcriptional levels. SFN could be potentially used as a single therapy or in combination with standard iron chelator drugs, but their drug–drug interaction should be determined through further experimental study. Considering the toxic nature of SFN at high concentrations, meticulous experiments should be done in vivo and in clinical settings prior to its utilization as an alternative treatment to alleviate iron overload-related conditions. Additionally, there are some challenges associated with the development of Nrf2 modulators, such as target specificity, pharmacodynamic responses, safety considerations, and determining the most appropriate indications [63]. The persistently high expression of Nrf2 can promote cancer development [83], which increases the challenges of using Nrf2 activators to treat iron overload.
Studies showed that drugs metabolized by the liver (substrates of cytochrome P450s, CYPs) interact with SFN, which may alter the enzymatic activities of CYPs, thus affecting the drugs’ effects and adverse reactions [84]. Another study has shown that SFN antagonistically interacts with furosemide (a loop diuretic), verapamil (a calcium channel blocker), and ketoprofen (a nonsteroidal anti-inflammatory drug) [85]. To the best of our knowledge, there is currently no evidence on the interaction between commercially available iron chelators and SFN. Therefore, further study is required to study the interactions between SFN and other iron chelators for clinical purposes.
5. ACKNOWLEDGMENTS
The results shown here are in part based upon data obtained from the NCBI Gene Expression Omnibus (GEO) profile database [http://www.ncbi.nlm.nih.gov/geo].
6. AUTHOR CONTRIBUTIONS
Conceptualization: FMS, SI; Investigation: APE, FMS; Writing—original draft preparation: APE, FMS; Writing—review and editing: SI; Funding acquisition: FMS, SI; Supervision: SI. All authors have contributed significantly to this study and approved the final manuscript.
7. FINANCIAL SUPPORT AND SPONSORSHIP
This study was supported by the Japan Society for the Promotion of Science [Grant 17K08581] (to SI), a Grant-in-Aid from Atma Jaya Catholic University of Indonesia (to FMS), and a Postdoctoral Grant from WCU UNDIP (to FMS).
8. CONFLICTS OF INTEREST
The authors report no financial or any other conflicts of interest in this work.
9. ETHICAL APPROVALS
This study does not involve experiments on animals or human subjects.
10. DATA AVAILABILITY
All the data is available with the authors and shall be provided upon request.
11. USE OF ARTIFICIAL INTELLIGENCE (AI)-ASSISTED TECHNOLOGY
The authors confirm that there was no use of artificial intelligence (AI)-assisted technology for assisting in the writing or editing of the manuscript and no images were manipulated using AI.
12. PUBLISHER’S NOTE
All claims expressed in this article are solely those of the authors and do not necessarily represent those of the publisher, the editors and the reviewers. This journal remains neutral with regard to jurisdictional claims in published institutional affiliation.
REFERENCES
1. Kasai S, Mimura J, Ozaki T, Itoh K. Emerging regulatory role of Nrf2 in iron, heme, and hemoglobin metabolism in physiology and disease. Front Vet Sci. 2018;5:242. [CrossRef]
2. Brissot P, Loréal O. Iron metabolism and related genetic diseases: a cleared land, keeping mysteries. J Hepatol. 2016;64(2):505–15. [CrossRef]
3. Sarosa H, Bahrudin U, Soemantri A, Muis SF, Arfian N, Hisatome I. The protective effect of azelnidipine for the prevention of heart fibrosis occurrence on Balb/c mice with iron overload. Bangladesh J Med Sci. 2020;19(2):223–8. [CrossRef]
4. Nienhuis AW, Nathan DG. Pathophysiology and clinical manifestations of the -Thalassemias. Cold Spring Harb Perspect Med. 2012;2:a011726. [CrossRef]
5. Pennell DJ, Udelson JE, Arai AE, Bozkurt B, Cohen AR, Galanello R, et al. Cardiovascular function and treatment in β-Thalassemia major. Circulation. 2013;128:281–308. [CrossRef]
6. Wilson JG, Maher JF, Lindquist JH, Grambow SC, Crook ED. Potential role of increased iron stores in diabetes. Am J Med Sci. 2003;325(6):332–9. [CrossRef]
7. Tantiworawit A, Kamolsripat T, Piriyakhuntorn P, Rattanathammethee T, Hantrakool S, Chai-Adisaksopha C, et al. Survival and causes of death in patients with alpha and beta-thalassemia in Northern Thailand. Ann Med. 2024;56(1):2338246. [CrossRef]
8. Budiwiyono I, AP P, Widyastiti NS, Hadian H, DK K. Correlation between ferritin levels with malondialdehyde and neutrophil lymphocyte ratio on iron overload. Indones J Clin Pathol Med Lab. 2021;27(2):147–51. [CrossRef]
9. van Raaij SEG, Srai SKS, Swinkels DW, van Swelm RPL. Iron uptake by ZIP8 and ZIP14 in human proximal tubular epithelial cells. BioMetals. 2019;32(2):211–26. [CrossRef]
10. Imam M, Zhang S, Ma J, Wang H, Wang F. Antioxidants mediate both iron homeostasis and oxidative stress. Nutrients. 2017;9(7):671. [CrossRef]
11. Ariningrum D, Adhipireno P, Suromo LB. Hyperferritinemia and oxidative stress in the kidney of beta thalassemia major. Bali Med J. 2019;8(2):596–601. [CrossRef]
12. Ray PD, Huang B-W, Tsuji Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell Signal. 2012;24(5):981–90. [CrossRef]
13. Siswanto FM, Okukawa K, Tamura A, Oguro A, Imaoka S. Hydrogen peroxide activates APE1/Ref-1 via NF-κB and Parkin: a role in liver cancer resistance to oxidative stress. Free Radic Res. 2023;57(3):223–38. [CrossRef]
14. Kontoghiorghes GJ. Advances on chelation and chelator metal complexes in medicine. Int J Mol Sci. 2020;21(7):2499. [CrossRef]
15. Entezari S, Haghi SM, Norouzkhani N, Sahebnazar B, Vosoughian F, Akbarzadeh D, et al. Iron chelators in treatment of iron overload. J Toxicol. 2022;2022:1–18. [CrossRef]
16. Kontoghiorghes GJ, Kontoghiorghe CN. Efficacy and safety of iron-chelation therapy with deferoxamine, deferiprone, and deferasirox for the treatment of iron-loaded patients with non-transfusion-dependent thalassemia syndromes. Drug Des Devel Ther. 2016;10:465–81. [CrossRef]
17. Umemura M, Kim J-H, Aoyama H, Hoshino Y, Fukumura H, Nakakaji R, et al. The iron chelating agent, deferoxamine detoxifies Fe(Salen)-induced cytotoxicity. J Pharmacol Sci. 2017;134(4):203–10. [CrossRef]
18. Bogdan AR, Miyazawa M, Hashimoto K, Tsuji Y. Regulators of iron homeostasis: new players in metabolism, cell death, and disease. Trends Biochem Sci. 2016;41(3):274–86. [CrossRef]
19. Gordan R, Wongjaikam S, Gwathmey JK, Chattipakorn N, Chattipakorn SC, Xie L-H. Involvement of cytosolic and mitochondrial iron in iron overload cardiomyopathy: an update. Heart Fail Rev. 2018;23(5):801–16. [CrossRef]
20. Baird SK, Kurz T, Brunk UT. Metallothionein protects against oxidative stress-induced lysosomal destabilization. Biochem J. 2006;394(Pt 1):275–83. [CrossRef]
21. Qiu Y, Zeng Y, Zhang C, Lv X, Ling Y, Si Y, et al. A ROS-responsive loaded desferoxamine (DFO) hydrogel system for traumatic brain injury therapy. Biomed Mater. 2024;19:025016. [CrossRef]
22. Amelia S, Nugroho T, Widyastiti N, Kholis F. The relation between types of iron chelators and feritin on osteocalcin of thalassemia patients with repeated transfusions. Diponegoro Med J. 2020;9(1):45–52. [CrossRef]
23. Hruby M, Martínez IIS, Stephan H, Pouckova P, Benes J, Stepanek P. Chelators for treatment of iron and copper overload: shift from low-molecular-weight compounds to polymers. Polymers (Basel). 2021;13(22):3969. [CrossRef]
24. Badawy SM, Palmblad J, Tricta F, Toiber Temin N, Fradette C, Lin L, et al. Rates of severe neutropenia and infection risk in patients treated with deferiprone: 28 years of data. Blood Adv. 2024. [CrossRef]
25. Kontoghiorghe C, Kolnagou A, Kontoghiorghes G. Phytochelators intended for clinical use in iron overload, other diseases of iron imbalance and free radical pathology. Molecules. 2015;20(11):20841–72. [CrossRef]
26. Wang X, Li Y, Han L, Li J, Liu C, Sun C. Role of flavonoids in the treatment of iron overload. Front Cell Dev Biol. 2021;9:685364. [CrossRef]
27. Alkharashi NAO, Periasamy VS, Athinarayanan J, Alshatwi AA. Sulforaphane alleviates cadmium-induced toxicity in human mesenchymal stem cells through POR and TNFSF10 genes expression. Biomed Pharmacother. 2019;115:108896. [CrossRef]
28. Alkharashi NAO, Periasamy VS, Athinarayanan J, Alshatwi AA. Sulforaphane mitigates cadmium-induced toxicity pattern in human peripheral blood lymphocytes and monocytes. Environ Toxicol Pharmacol. 2017;55:223–39. [CrossRef]
29. Kubo E, Chhunchha B, Singh P, Sasaki H, Singh DP. Sulforaphane reactivates cellular antioxidant defense by inducing Nrf2/ARE/Prdx6 activity during aging and oxidative stress. Sci Rep. 2017;7:14130. [CrossRef]
30. Schepici G, Bramanti P, Mazzon E. Efficacy of sulforaphane in neurodegenerative diseases. Int J Mol Sci. 2020;21(22):8637. [CrossRef]
31. Houghton CA, Fassett RG, Coombes JS. Sulforaphane and other nutrigenomic Nrf2 activators: can the clinician’s expectation be matched by the reality? Oxid Med Cell Longev. 2016;2016:1–17. [CrossRef]
32. Siswanto FM, Firmasyah RD, Handayani MDN, Oguro A, Imaoka S. Nrf2 regulates the expression of CYP2D6 by inhibiting the activity of Krüppel-Like Factor 9 (KLF9). Curr Drug Metab. 2023;24(9):667–81. [CrossRef]
33. Ross D, Siegel D. NQO1 in protection against oxidative stress. Curr Opin Toxicol. 2018;7:67–72. [CrossRef]
34. Loboda A, Damulewicz M, Pyza E, Jozkowicz A, Dulak J. Role of Nrf2/HO-1 system in development, oxidative stress response and diseases: an evolutionarily conserved mechanism. Cell Mol Life Sci. 2016;73:3221–47. [CrossRef]
35. Ma Q. Role of Nrf2 in oxidative stress and toxicity. Annu Rev Pharmacol Toxicol. 2013;53:401–26. [CrossRef]
36. Wen Z, Liu W, Li X, Chen W, Liu Z, Wen J, et al. A protective role of the NRF2-Keap1 pathway in maintaining intestinal barrier function. Oxid Med Cell Longev. 2019;2019:1–7. [CrossRef]
37. Siswanto FM, Oguro A, Arase S, Imaoka S. WDR23 regulates the expression of Nrf2-driven drug-metabolizing enzymes. Drug Metab Pharmacokinet. 2020;35(5):441–55. [CrossRef]
38. Taher AT, Saliba AN. Iron overload in thalassemia: different organs at different rates. Hematology. 2017;2017(1):265–71. [CrossRef]
39. Siswanto FM, Sakuma R, Oguro A, Imaoka S. Chlorogenic acid activates Nrf2/SKN-1 and prolongs the lifespan of Caenorhabditis elegans via the Akt-FOXO3/DAF16a-DDB1 pathway and activation of DAF16f. J Gerontol Ser A. 2022;77(8):1503–16. [CrossRef]
40. Eruslanov E, Kusmartsev S. Identification of ROS using oxidized DCFDA and flow-cytometry. Methods Mol Biol. 2010:594:57–72. [CrossRef]
41. Siswanto FM, Tamura A, Sakuma R, Imaoka S. Yeast β -glucan increases etoposide sensitivity in lung cancer cell line A549 by suppressing nuclear factor erythroid 2-related factor 2 via the noncanonical nuclear factor kappa B pathway. Mol Pharmacol. 2022;101(4):257–73. [CrossRef]
42. Baba K, Morimoto H, Imaoka S. Seven in absentia homolog 2 (Siah2) protein is a regulator of NF-E2-related factor 2 (Nrf2). J Biol Chem. 2013;288:18393–405. [CrossRef]
43. Barrett T, Wilhite SE, Ledoux P, Evangelista C, Kim IF, Tomashevsky M, et al. NCBI GEO: archive for functional genomics data sets—update. Nucleic Acids Res. 2012;41:D991–5. [CrossRef]
44. Endale HT, Tesfaye W, Mengstie TA. ROS induced lipid peroxidation and their role in ferroptosis. Front Cell Dev Biol. 2023;11:1226044. [CrossRef]
45. Zhenxiong L, Dou W, Zheng Y, Wen Q, Qin M, Wang X, et al. Curcumin upregulates Nrf2 nuclear translocation and protects rat hepatic stellate cells against oxidative stress. Mol Med Rep. 2016;13(2):1717–24. [CrossRef]
46. Siswanto FM, Oguro A, Imaoka S. Sp1 is a substrate of Keap1 and regulates the activity of CRL4A(WDR23) ubiquitin ligase toward Nrf2. J Biol Chem. 2021;296:100704. [CrossRef]
47. Liu Y, Liu P, Wang Q, Sun F, Liu F. Sulforaphane attenuates H?O?-induced oxidant stress in human trabecular meshwork cells (HTMCs) via the phosphatidylinositol 3-kinase (PI3K)/serine/threonine kinase (Akt)-mediated factor-E2-related factor 2 (Nrf2) signaling activation. Med Sci Monit. 2019;25:811–8. [CrossRef]
48. Jang CH, Oh J, Kim J-S. Nrf2?activating phytochemicals, sulforaphane and licochalcone A, stimulate cell growth?regulating kinases in HCT116 human colorectal cancer cells. FASEB J. 2020;34(S1):1. [CrossRef]
49. Li Y, Ouyang Q, Chen Z, Chen W, Zhang B, Zhang S, et al. Intracellular labile iron is a key regulator of hepcidin expression and iron metabolism. Hepatol Int. 2023;17(3):636–47. [CrossRef]
50. Ong AJS, Bladen CE, Tigani TA, Karamalakis AP, Evason KJ, Brown KK, et al. The KEAP1–NRF2 pathway regulates TFEB/TFE3-dependent lysosomal biogenesis. Proc Natl Acad Sci USA. 2023;120(22):e2217425120. [CrossRef]
51. Gross-Steinmeyer K, Stapleton PL, Tracy JH, Bammler TK, Strom SC, Eaton DL. Sulforaphane- and phenethyl isothiocyanate–induced inhibition of aflatoxin B1–mediated genotoxicity in human hepatocytes: role of GSTM1 genotype and CYP3A4 gene expression. Toxicol Sci. 2010;116(2):422–32. [CrossRef]
52. Abbaspour N, Hurrell R, Kelishadi R. Review on iron and its importance for human health. J Res Med Sci. 2014;19(2):164–74. [CrossRef]
53. Hsu CC, Senussi NH, Fertrin KY, Kowdley KV. Iron overload disorders. Hepatol Commun. 2022;6(8):1842–54. [CrossRef]
54. Su L-J, Zhang J-H, Gomez H, Murugan R, Hong X, Xu D, et al. Reactive oxygen species-induced lipid peroxidation in apoptosis, autophagy, and ferroptosis. Oxid Med Cell Longev. 2019;2019:1–13. [CrossRef]
55. Pinto V, Forni G. Management of iron overload in beta-thalassemia patients: clinical practice update based on case series. Int J Mol Sci. 2020;21(22):8771. [CrossRef]
56. Siswanto FM, Mitsuoka Y, Nakamura M, Oguro A, Imaoka S. Nrf2 and Parkin-Hsc70 regulate the expression and protein stability of p62/SQSTM1 under hypoxia. Sci Rep. 2022;12:21265. [CrossRef]
57. Suzuki T, Yamamoto M. Molecular basis of the Keap1–Nrf2 system. Free Radic Biol Med. 2015;88(Pt B):93–100. [CrossRef]
58. Tonelli C, Chio IIC, Tuveson DA. Transcriptional regulation by Nrf2. Antioxid Redox Signal. 2018;29(17):1727–45. [CrossRef]
59. Suprihatin T, Rahayu S, Rifa’i M, Widyarti S. Distribution of Nrf2 and GST Enzyme in premenopausal white rat ovarian granulosa cells after administration of turmeric powder. Bul Anat Dan Fisiol. 2021;6(1):66–73.
60. Kerins MJ, Ooi A. The roles of NRF2 in modulating cellular iron homeostasis. Antioxid Redox Signal. 2018;29(17):1756–73. [CrossRef]
61. Lim PJ, Duarte TL, Arezes J, Garcia-Santos D, Hamdi A, Pasricha S-R, et al. Nrf2 controls iron homoeostasis in haemochromatosis and thalassaemia via Bmp6 and hepcidin. Nat Metab. 2019;1(5):519–31. [CrossRef]
62. Dodson M, de la Vega MR, Cholanians AB, Schmidlin CJ, Chapman E, Zhang DD. Modulating Nrf2 in disease: timing is everything. Annu Rev Pharmacol Toxicol. 2019;59:555–75. [CrossRef]
63. Dinkova-Kostova AT, Copple IM. Advances and challenges in therapeutic targeting of NRF2. Trends Pharmacol Sci. 2023;44(3):137–49. [CrossRef]
64. Otoo RA, Allen AR. Sulforaphane’s Multifaceted potential: from neuroprotection to anticancer action. Molecules. 2023;28(19):6902. [CrossRef]
65. Pouremamali F, Pouremamali A, Dadashpour M, Soozangar N, Jeddi F. An update of Nrf2 activators and inhibitors in cancer prevention/promotion. Cell Commun Signal. 2022;20(1):100. [CrossRef]
66. Thangapandiyan S, Ramesh M, Miltonprabu S, Hema T, Jothi GB, Nandhini V. Sulforaphane potentially attenuates arsenic-induced nephrotoxicity via the PI3K/Akt/Nrf2 pathway in albino Wistar rats. Environ Sci Pollut Res. 2019;26(12):12247–63. [CrossRef]
67. Baird L, Yamamoto M. The molecular mechanisms regulating the KEAP1-NRF2 pathway. Mol Cell Biol. 2020;40(13):e00099–20. [CrossRef]
68. Chau L-Y. Heme oxygenase-1: emerging target of cancer therapy. J Biomed Sci. 2015;22(1):22. [CrossRef]
69. Jiang T, Cheng H, Su J, Wang X, Wang Q, Chu J, et al. Gastrodin protects against glutamate-induced ferroptosis in HT-22 cells through Nrf2/HO-1 signaling pathway. Toxicol Vitr. 2020;62:104715. [CrossRef]
70. Ma H, Wang X, Zhang W, Li H, Zhao W, Sun J, et al. Melatonin suppresses ferroptosis induced by high glucose via activation of the Nrf2/HO-1 signaling pathway in type 2 diabetic osteoporosis. Oxid Med Cell Longev. 2020;2020:1–18. [CrossRef]
71. Wei N, Lu T, Yang L, Dong Y, Liu X. Lipoxin A4 protects primary spinal cord neurons from Erastin?induced ferroptosis by activating the Akt / Nrf2 / HO?1 signaling pathway. FEBS Open Bio. 2021;11(8):2118–26. [CrossRef]
72. Ling X-B, Wei H-W, Wang J, Kong Y-Q, Wu Y-Y, Guo J-L, et al. Mammalian metallothionein-2A and oxidative stress. Int J Mol Sci. 2016;17(19):1483. [CrossRef]
73. Houessinon A, François C, Sauzay C, Louandre C, Mongelard G, Godin C, et al. Metallothionein-1 as a biomarker of altered redox metabolism in hepatocellular carcinoma cells exposed to sorafenib. Mol Cancer. 2016;15(1):38. [CrossRef]
74. Sakulsak N. Metallothionein: an overview on its metal homeostatic regulation in mammals. Int J Morphol. 2012;30(3):1007–12. [CrossRef]
75. Kr??el A, Maret W. Different redox states of metallothionein/thionein in biological tissue. Biochem J. 2007;402(Pt 3):551–8. [CrossRef]
76. Dong G, Chen H, Qi M, Dou Y, Wang Q. Balance between metallothionein and metal response element binding transcription factor 1 is mediated by zinc ions (Review). Mol Med Rep. 2015;11(3):1582–6. [CrossRef]
77. Wyrich M, Ohlig H, Wessolly M, Mairinger E, Steinborn J, Brcic L, et al. Induction of metallothionein expression by supplementation of zinc induces resistance against platinum-based treatment in malignant pleural mesothelioma. Transl Cancer Res. 2023;12(8):1929–36. [CrossRef]
78. Kooncumchoo P, Sharma S, Porter J, Govitrapong P, Ebadi M. Coenzyme Q(10) provides neuroprotection in iron-induced apoptosis in dopaminergic neurons. J Mol Neurosci. 2006;28(2):125–42. [CrossRef]
79. Vignesh KS, Deepe Jr. G. Metallothioneins: emerging modulators in immunity and infection. Int J Mol Sci. 2017;18(10):2197. [CrossRef]
80. Gu J, Cheng Y, Wu H, Kong L, Wang S, Xu Z, et al. Metallothionein is downstream of Nrf2 and partially mediates sulforaphane prevention of diabetic cardiomyopathy. Diabetes. 2017;66(2):529–42. [CrossRef]
81. Wu H, Kong L, Cheng Y, Zhang Z, Wang Y, Luo M, et al. Metallothionein plays a prominent role in the prevention of diabetic nephropathy by sulforaphane via up-regulation of Nrf2. Free Radic Biol Med. 2015;89:431–42. [CrossRef]
82. Yeh C-T, Yen G-C. Effect of sulforaphane on metallothionein expression and induction of apoptosis in human hepatoma HepG2 cells. Carcinogenesis. 2005;26(12):2138–48. [CrossRef]
83. Lin L, Wu Q, Lu F, Lei J, Zhou Y, Liu Y, et al. Nrf2 signaling pathway: current status and potential therapeutic targetable role in human cancers. Front Oncol. 2023;13:1184079. [CrossRef]
84. Srovnalova A, Vanduchova A, Svecarova M, Anzenbacherova E, Tomankova V, Anzenbacher P, et al. Effects of sulforaphane and its S- and R-enantiomers on the expression and activities of human drug-metabolizing cytochromes P450. J Funct Foods. 2015;14:487–501. [CrossRef]
85. Lubelska K, Milczarek M, Modzelewska K, Krzyszto?-Russjan J, Fronczyk K, Wiktorska K. Interactions between drugs and sulforaphane modulate the drug metabolism enzymatic system. Pharmacol Rep. 2012;64(5):1243–52. [CrossRef]