
1. INTRODUCTION
Vaccines have emerged as a highly efficacious and economically viable medical strategy, resulting in the preservation of countless lives [1]. Mass vaccination campaigns have led to the eradication of pathogens such as the smallpox virus, which has inflicted significant casualties [2]. Nevertheless, despite remarkable achievements have been made, the development of efficacious vaccines remains incomplete for intricate pathogens responsible for severe diseases, such as malaria, HIV/AIDS, and tuberculosis [3].
Generally, vaccines have been formulated using whole pathogens, which involve either attenuated strains (classified as live-attenuated vaccines) or inactivated variants (inactivated through heat or formalin treatment) [1], as summarized in Table 1. However, the persistence of these traditional methodologies to engineer whole-pathogen vaccines presents several complex technical obstacles [4]. Moreover, such vaccines carry substantial safety concerns, including the potential to revert to virulent states, inducing severe reactions in individuals with compromised immune systems, and triggering undesirable outcomes such as allergic or autoimmune responses [5,6].
Table 1: Different types of vaccines [4].
| Vaccine platform | Vaccine component | Vaccine feature |
|---|---|---|
| Inactivated vaccine | Viruses or bacteria are cultured in vitro and inactivated. | Immunogenicity is lower than live attenuated vaccine. Immunity is short-lived, requiring multiple vaccinations. |
| Live attenuated vaccine | Viruses or bacteria are obtained by reverse genetics or adaption. | Attenuated pathogens mimic live pathogen infection but are weakly pathogenic. Poor stability and low safety may cause virulence re-emergence. |
| Subunit vaccine | Antigen-specific proteins or peptides are expressed by cell-expressing systems. | Subunit vaccines offer better safety, purity, scalability, suitability, and stability for immunocompromised people. |
| DNA vaccine | Antigens are encoded by a recombinant plasmid. | Plasmid DNA is economical, safe, and stable. However, delivery to the nucleus can be difficult, leading to insertional mutagenesis. |
| mRNA vaccine | mRNA encodes protein antigens encapsulated by vectors. | mRNA does not enter the nucleus and does not alter the genome. It has an immune-activating effect, but safety and stability are issues. |
| Viral vector vaccine | Modified viruses with weakened replication and antigen-encoding genes. | Viral vector has an immune-stimulating effect. Its immune and safety effects need improvement. |
| VLP vaccine | Structural proteins form vaccine immunogens. | Vaccines similar to actual virions but without the virus genome, are safe, stable, structured, appropriately sized, and modifiable. |
Subunit vaccines, which consist of specific antigenic components of pathogens, offer a safer alternative to whole-pathogen vaccines and hold promise for addressing vaccine challenges [7]. Notably, subunit vaccines have gained traction because of their safety profile and efficacy against diverse infectious diseases such as hepatitis B, diphtheria, shingles, tetanus, and cervical cancer [8]. This approach also has significance in the context of COVID-19, where numerous subunit vaccine candidates are in clinical and pre-clinical stages. Notably, the NVX-COV2373 COVID-19 subunit vaccine demonstrated comparable CD4+ T-cell memory responses and neutralizing antibodies to those of mRNA vaccines [9]. In addition, subunit vaccines targeting well-defined epitopes hold potential in cancer immunotherapy, as exemplified by ongoing clinical trials for tumor neoantigen-based subunit vaccines (ChiCTR2000029301 and ChiCTR1800016628) and the 9-valent human papillomaviruses (HPV) subunit vaccine (NCT05266898) [10]. The landscape includes 103 neoantigen-based subunit vaccine clinical studies registered at ClinicalTrials.gov [10].
Despite their advantages, subunit vaccines exhibit limitations in generating robust immune responses compared to whole pathogens [7]. Recent progress, notably in identifying immunostimulatory elements [11] and optimizing vaccine delivery platforms [12], has enabled the rational design of potent subunit vaccines capable of conferring enduring protective immunity [13]. However, a key challenge in subunit vaccine development is the selection of efficient delivery systems with minimal or no toxicity.
This review aims to comprehensively examine the recent advances in subunit vaccine delivery systems, including polymer-based, lipid-based, micelle-based, phage-based, hydrogel-based, inorganic-based, and emulsion-based vaccine delivery systems. Their advantages, disadvantages, research gaps, and future directions were also discussed.
2. SUBUNIT VACCINE-INDUCED IMMUNITY PATHWAYS
2.1. Innate Immune Responses
The innate immune system has evolved mechanisms to identify evolutionarily conserved pathogen-associated molecular patterns (PAMPs), facilitating recognition through pattern recognition receptors (PRRs) found on various innate immune cells, such as neutrophils, mast cells, macrophages, and dendritic cells (DCs) [14]. The engagement of receptors triggers innate immune cell activation, prompting an inflammatory response that promotes the migration of immune cells, including antigen-presenting cells (APCs) and neutrophils, from circulation to infection sites [Figure 1] [15]. Distributed widely, DCs play a pivotal role as professional APCs, strategically positioned on lymphoid organs and mucosal surfaces [16]. PRRs enable migratory and tissue-resident DCs to effectively sense pathogens, internalize antigens through phagocytosis and micropinocytosis, and undergo maturation [17]. Maturation results in diminished antigen uptake heightened antigen-processing machinery expression, and the surface translocation of antigen peptide-bound major histocompatibility complex (MHC) molecules [18]. To trigger adaptive immune responses, DC maturation facilitates clonal expansion and differentiation of antigen-specific naive T cells into effector T cells, thus contributing to immune defense [19].
 | Figure 1: Innate and adaptive immune response upon vaccination. [Click here to view] |
DC maturation prompts alterations in adhesion molecules and chemokine receptor expression, which enable migration to peripheral lymphoid organs that are crucial for initiating adaptive immunity [Figure 1] [18]. Whole pathogen vaccines, typified by live-attenuated variants, are characterized by multiple PAMPs that facilitate robust recognition by PRRs and potent innate immune responses [20]. In contrast, (non-viral) subunit vaccines that comprise specific antigens lack inherent PAMPs, necessitating the inclusion of adjuvants or immunostimulators to induce robust adaptive immune responses [21].
2.2. Adaptive Immune Responses
Adaptive immune responses are initiated in the peripheral lymphoid organs after antigen presentation by mature DCs, stemming from the innate immune reaction [Figure 1] [22]. Essential to the onset of adaptive immunity, T-cell activation hinges on encountering pathogen antigens presented by mature DCs. Notably, three signals underpin naive T-cell activation: Signal 1 involves T-cell receptor (TCR) interaction with antigen peptide-MHC, signal 2 encompasses CD28-B7 co-stimulation, and signal 3 orchestrated by DC-secreted cytokines, directs T-cell differentiation into effector subsets [23]. Naive T cells will then differentiate into CD4+ and CD8+ T cells based on the presence of TCR coreceptors. CD8+ T cells, which recognize MHC-I-presented antigens, differentiate into cytotoxic T lymphocytes (CTLs) that are vital for intracellular pathogen defense [Figure 1] [24]. Intracellular pathogen infection triggers MHC-I-presented cytosolic antigen peptides, which render CTLs capable of identifying and eliminating infected cells [24]. In contrast, inactivated or subunit vaccines, which are extracellular antigens, predominantly employ MHC class II presentation by the APCs [25]. Nevertheless, selected extracellular antigens, potentially virus-like particle (VLP) vaccines, may undergo MHC-I-mediated “cross-presentation,” activating CD8+ T cells [Figure 1] [24]. Enhancing cross-presentation efficacy, particularly against intracellular pathogens, is a key focus in optimizing subunit vaccines [26].
In contrast, CD4+ T cells recognized the antigenic peptides displayed by MHC-II [Figure 1]. In contrast to CD8+ T cells and CD4+ T cells exhibit diverse effector differentiations, termed helper T (Th) cells, each governed by distinct cytokines [27]. Th1 effectors, which are driven by interferon γ (IFNγ) and interleukin 12 (IL-12), combat intracellular pathogens. Th2 cells, activated by IL4 and IL2, primarily target the extracellular parasites. Th17 cells, induced by IL21, IL6, IL23, and transforming growth factor-β, target extracellular bacteria and fungi [28]. Tfh cells, localized in lymphoid tissue follicles, play a pivotal role in fostering an antigen-specific humoral immune response [27].
B cells, which act as precursors to antibody-secreting plasma cells, play crucial roles in humoral immunity by protecting against extracellular pathogens. The efficient activation of B cells typically requires the assistance of effector Th cells [Figure 1]. Within lymphoid tissues, B and T cells occupy separate domains (B-cell zones and T-cell zones) [29]. Naive T cells arriving in lymphoid tissues interact with activated DCs and differentiate into Th cells [30]. Naive B cells will then enter the T-cell zone, engage with Th cells, and subsequently move to the B-cell zone [29]. Activation of B cells typically relies on dual signals: Pathogen antigen interaction with surface immunoglobulin (or B-cell receptor), leading to internalization and pathogen peptide presentation through MHC-II (signal 1) [30]. Recognition of MHC-II-bound peptides provides a second signal involving CD40-CD40L interaction and Th cell-secreted cytokines. Subsequently, activated B cells move to the lymphoid follicles, establishing germinal centers. Here, B cells undergo somatic hypermutation and affinity-driven selection [29]. Emerging from germinal centers, some B cells develop into either memory B cells in circulation or antibody-secreting plasma cells [30].
3. SUBUNIT VACCINE DELIVERY AND ADMINISTRATION
Effective vaccine delivery approaches are essential for triggering appropriate immune responses and establishing long-lasting immunological memory, which is crucial for preventing future infections [31]. The efficacy of immunization and vaccine delivery is heavily contingent on the chosen route of administration because suboptimal administration may compromise vaccine effectiveness [32]. Ideally, administering vaccines in proximity to lymph nodes or lymphatic vessels amplifies immune responses, although this is modulated by antigenic epitopes and adjuvant functionality [33]. Two principal avenues have been explored for optimizing vaccine delivery: Parenteral and non-parenteral.
The parenteral vaccine delivery approach involves vaccine administration bypassing the gastrointestinal tract and is typically administered through injection or infusion using hypodermic needles. Common examples are the intradermal, subcutaneous, and intramuscular routes [34]. This widely adopted strategy tailors vaccine placement according to anatomical site characteristics: The epidermis, hypodermis, and dermis [35]. Optimal site selection is critical for efficient vaccine delivery. In adults, the deltoid region serves as the choice for intradermal and intramuscular administration, while the outer triceps area is preferred for subcutaneous delivery. Conversely, the anterolateral thigh region becomes significant in toddlers and infants [36]. Precision in needle selection, accounting for tissue thickness, muscle size, and diverse factors contingent on age, body mass, and sex, is imperative for this approach [37].
Although needle-based injection devices have offered promising vaccine delivery solutions, drawbacks such as needle stick injuries and cost inefficiencies have emerged as significant concerns [38]. Needle-free devices have been developed to enhance efficiency and alleviate pain during vaccine administration [38]. Various non-parenteral needle-free strategies have emerged, including powder, liquid, and projectile approaches as well as jet injectors such as spring-loaded, battery-powered, and gas-powered devices [37]. Nevertheless, these methods have limitations in eliciting mucosal immunity [38]. Conversely, non-parenteral vaccine strategies leverage live or inactivated antigen molecules to induce antigen-mediated immune responses. This approach includes oral, intranasal, and transcutaneous routes of administration [36].
4. DELIVERY SYSTEMS FOR SUBUNIT VACCINES
4.1. Polymer-based Vaccine Delivery
Recently, there has been a surge in interest in polymers as potential antigen carriers. These adaptable molecules have dual roles as both delivery systems and immunostimulants in vaccine formulations [Figure 2]. Polymers with inherent immunostimulatory attributes engage immune cell receptors, direct antigen delivery to specific uptake sites, and initiate distinct immune pathways. Typically, immunostimulants are co-administered with antigens, either through physical mixing or chemical linkage, to evoke targeted and tailored immune responses [39,40]. These polymer-based adjuvants are discussed below.
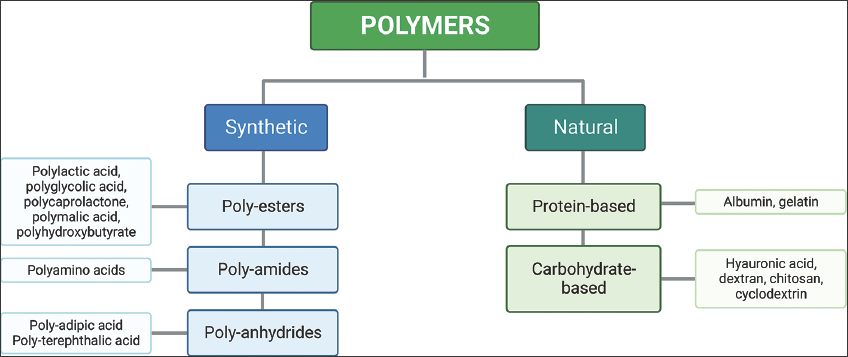 | Figure 2: Polymers used in vaccine delivery. [Click here to view] |
4.1.1. Polysaccharide polymers
4.1.1.1. Chitosan
Chitosan, an eco-friendly and biocompatible polymer, has distinctive attributes such as mucoadhesion and cationic properties stemming from its abundant free amine groups, which can form salts in low-pH conditions [41]. The hydroxyl groups allow for facile attachment or modification of peptides or proteins [42]. The immunostimulatory effects of chitosan include augmented cellular and humoral immune responses [43]. By interacting with diverse receptors on APCs, including dectin-1, mannose receptors, leukotriene B4, and toll-like receptor (TLR)-2, this polymer and its derivatives further underscore their potential for immune modulation [44].
Chitosan is an exceptional adjuvant for mucosal delivery, due to mucoadhesion and ability to induce junction openings, thus facilitating the paracellular passage of vaccine antigens [45]. The cationic nature of chitosan promotes intensified cellular interactions with anionic epithelial cells, extending the presence of antigens within the nasal cavity [46]. By leveraging charge-based interactions, influenza A virus matrix protein 1 (M1, 100 μg) co-administered with chitosan through intranasal delivery resulted in elevated immunoglobulin (Ig)G and IgA titers against the virus in mice [47].
4.1.1.2. Alginate
Alginate, a bioadhesive polysaccharide polymer renowned for its anionic character, has gained prominence as a drug delivery system due to its capacity for gastric contractions and intestinal cargo release. Recent efforts have extended the applicability of alginate to vaccine delivery. The research underscores the adjuvant potential of alginate as it stimulates monocytes/macrophages [48]. Particularly noteworthy is the role of alginate in facilitating site-specific vaccine antigen delivery to mucosal tissues. Its inclusion in formulations enhances phagocytosis and bolsters formulation adhesion to DCs, amplifying its influence [49].
Alginate has emerged as a promising contender for subunit vaccine delivery, demonstrating versatility in formulations such as conjugates, nanogels, and microparticles. An illustrative case involves the linkage of Pseudomonas aeruginosa-derived peptide antigens (peptide294 and peptide176) with alginate [49]. Subcutaneous administration of the peptide294-alginate conjugate emulsified with incomplete Freund’s adjuvant (IFA) produced robust levels of protective and opsonophagocytic antibodies in mice. In contrast, peptide294 administered with IFA alone failed to trigger a marked humoral response [49].
4.1.1.3. Hyaluronic acid (HA)
HA, also termed hyaluronan, is a linear mucopolysaccharide with several biomedical applications [50]. HA’s exceptional hydrophilicity of HA establishes it as nature’s most hydrophilic polymer [51]. A notable trait of HA is its non-immunogenic and non-antigenic nature, attributed to its extensively conserved structure across species. This innate polymer is widespread among prokaryotes and eukaryotes and is widely distributed within the extracellular and pericellular matrix, as well as intracellularly [51].
HA has emerged as a key player in transdermal immunization because of its skin-hydrating properties and facilitation of skin surface absorption and permeation. In collaboration with antigenic peptides, HA enables deep delivery into the skin layers, capitalizing on its skin-penetrating and hygroscopic characteristics [52]. HA interacts with dermal DCs and epidermal Langerhans cells through HA receptors and immune-cell-present TLRs. Intriguingly, low-molecular-weight HA acts as an intrinsic danger signal, activating TLR2- and TLR4-mediated transduction pathways [53]. Moreover, it exerts immunostimulatory effects by fostering chemokine and cytokine production. TLR2 and TLR4 pathway activation through low-MW HA reinforces the skin’s self-defense mechanisms, culminating in β-defensin 2 production [54]. In a study targeting transdermal immunotherapy for Duchenne muscular dystrophy, myostatin fragment (MstnF)-derived antigenic peptides, namely MstnF and scrMstnF, were conjugated with HA [52]. Transdermal immunization of mdx mice with HA-MstnF conjugate showed a substantial surge in myostatin-specific antibody titers. This translates to remarkable enhancements in skeletal muscle biochemistry and pathology, along with notable functional improvement [52].
4.1.1.4. Dextran
Dextran is an intricately branched polysaccharide with notable water solubility and controlled degradation. Various dextran derivatives have garnered attention owing to their adjuvant properties. In particular, dextran sulfate presents compelling prospects as a material for controlled pharmaceutical release. The notable charge density of dextran sulfate, arising from the high sulfate-to-glucosyl ratio, facilitates the enhanced loading of positively charged molecules [55].
Conjugation of dextran with bovine serum albumin has demonstrated remarkable efficacy in eliciting robust and sustained antibody responses in mice, even in the absence of supplementary adjuvants. Remarkably, detectable antibody titers were achieved at a mere 10 μg dosage, with a clear dose-dependent elevation in titers at higher dosages. The molecular weight of dextran emerged as a pivotal determinant of antibody titers, with dextran within the 500–2000 kDa range proving essential, whereas 70 kDa dextran failed to trigger detectable antibody production [56]. Moreover, dextran has emerged as a versatile platform for the conjugation of TLR7 agonist 1V209 and CpG oligodeoxynucleotide (CpG ODN) adjuvant, yielding heightened targeting precision and enhanced immunostimulatory profiles for these adjuvants [57]. These findings underscore the adaptable nature of dextran for enhancing adjuvant functionality, thus broadening its potential applications in the field [56].
4.1.1.5. Carrageenan
Carrageenan from red seaweed has emerged as a promising adjuvant for peptide vaccines, attracting notable attention in recent research. The distinctive anionic character of carrageenan stems from its hemisulfate ester groups owing to its unique attributes. The structural foundation of carrageenan involves a polymer chain consisting of hemisulfated galactose and 3,6-anhydrogalactose residues. Based on the positioning and arrangement of ester sulfate groups within recurring galactose monomers, carrageenans can be classified into three main types: Kappa (κ-), lambda (λ-), and iota (ι-) carrageenans [58].
The use of carrageenan as a delivery system stems from its unique ability to provoke antigen-specific immunity and exhibit antitumor effects. In an experimental context involving mice immunized with a blend of carrageenan and an E7 protein-derived peptide from HPV-16, carrageenan notably amplified immune responses directed at the E7 antigen through TLR4 pathway activation [59]. Crucially, the augmented immune reaction induced by carrageenan was similar to that induced by established TLR4 ligands, including monophosphoryl lipid A and dextran [59]. This discovery underscores the promising adjuvant characteristics of carrageenan, reaffirming its potential to enhance immune responses similar to those of established immunostimulants.
4.1.2. Polyesters
4.1.2.1. Poly(ε-caprolactone) (PCL)
PCL is a polyester with inherent biodegradability because its ester linkages hydrolyze under physiological conditions. Notably, PCL’s degradation rate of PCL is slower than that of polylactide (PLA) polymers [60]. A distinctive trait of PCL is its ability to avert acidic environment generation upon dissolution, which is in contrast to other polyesters such as poly(lactic-co-glycolic acid) (PLGA). This feature is particularly advantageous as it prevents potential harm to the antigenicity of loaded proteins or peptides. PCL biodegradability and safety have led to the Food and Drug Administration’s approval of long-term implantable devices. Moreover, PCL’s attributes of PCL include hydrophobicity, biocompatibility, and cost-effectiveness, thereby positioning it as a versatile polymer in various applications [61].
PCL has emerged as a prominent choice for sustained-release antigen delivery, negating the requirement for prime-boosting, owing to its naturally slow biodegradation in vivo. This dual impact allowed the initial surface-released antigen from the PCL microspheres to act as a priming dose. Prolonged antigen release, facilitated by diffusion or microsphere degradation, serves as a boosting effect. An illustrative study explored immunogenicity using 23 μm PCL microspheres loaded with ovalbumin (OVA) [62]. Administration of these microspheres induced elevated IgG responses compared with standalone OVA, although notably lower than OVA paired with complete Freund’s adjuvant.
4.1.2.2. PLGA
PLGA is a copolymer in which glycolic or lactic acid monomers are linked by ester bonds. PLGA, as a delivery platform for vaccines, has considerable potential because of several crucial factors. Notably, controlled degradation of PLGA particles before their uptake by APCs enhances their desirability. Moreover, the inherent non-toxic nature and ability to facilitate precise antigen release [63] contribute significantly to its utility in vaccine delivery.
The degradation mechanism of PLGA involves bulk erosion, which allows water to infiltrate the polymer matrix. This triggers ester bond hydrolysis and the regeneration of glycolic acid and lactic acid monomers. Importantly, these by-products have minimal toxic effects, owing to their involvement in physiological pathways. Simultaneously, this degradation leads to heightened matrix porosity, facilitating gradual antigen release as degradation proceeds [64].
In an experimental study, PLGA microspheres incorporating adjuvant calcium phosphate gel were utilized to encapsulate the OVA antigen. Nasal immunization with these microspheres, sized 7 μm and carrying a −22 mV surface charge, predominantly induced IgG1 titers in both the serum and local mucosa. These IgG1 titers signify a Th2 immune response and were found comparable to those induced by administering OVA along with cholera toxin subunit B (CTB). Thus, the PLGA formulation exhibited adjuvant-like properties similar to those of the established mucosal adjuvant CTB [65]. These results highlight the potential of PLGA microspheres combined with calcium phosphate gel as a promising approach for mucosal immunization.
4.1.3. Polyglutamic acid (PGA)
PGA is a non-toxic, biocompatible, and biodegradable polymer composed of repetitive glutamic acid units. This versatile polymer has two forms, γ-PGA and α-PGA, with distinct linkages: α-carboxylic acids in α-PGA and γ-carboxylic acids in γ-PGA. Notably, α-PGA is typically synthesized chemically, whereas γ-PGA is biosynthesized by Bacillus species [66].
PGA nanoparticles have emerged as effective vaccine carriers, facilitating targeted delivery of antigenic proteins to APCs and generating robust immune responses. One approach involves grafting γ-PGA polymer with L-phenylalanine ethyl ester (L-PAE) [67]. These γ-PGA-L-PAE NPs efficiently encapsulated OVA, exhibited effective uptake by immature DCs, and promoted DC maturation. Compared to OVA alone, OVA-loaded γ-PGA-L-PAE NPs demonstrated enhanced efficiency in inducing cellular CTL responses, comparable to OVA with complete Freund’s adjuvant [68]. In a separate study, immunization with γ-PGA-L-PAE NPs coated with the CD8+ T-cell epitope listeriolysin O (LLO) peptide (VAYGRQVYLKLS) resulted in a remarkable 11-day survival period post-challenge and a significant improvement in mice receiving PBS or LLO alone [69]. These findings underscore the potential of modified γ-PGA NPs as a promising platform for enhancing vaccine efficacy and immune response modulation.
4.1.4. Polyacrylates
Poly(methyl methacrylate) (PMMA), a synthetic homopolymer derived from methyl methacrylate monomers, has attracted significant attention in the biomedical field owing to its remarkable biocompatibility. Although inherently hydrophobic, PMMA exhibits a modest increase in hydrophilicity upon contact with water. Its well-established biocompatibility and safety profile have led to its application in various biomedical contexts, such as implant materials for total hip replacements [70].
The potential of PMMA as a vaccine delivery system was initially demonstrated by Kreuter and Speiser. In their work, PMMA was shown to enhance immune responses when combined with inactivated influenza virus, highlighting its adjuvant capabilities [71]. In addition, PMMA microspheres have been observed to be absorbed by gut-associated lymphoid tissues, suggesting their potential for oral vaccine delivery [72]. Despite being non-biodegradable, PMMA has been investigated as a vaccine delivery strategy. For example, core-shell nanoparticles incorporating anionic cores and Eudragit-derived shells featuring adsorbed HIV Tat protein (220 nm in size) were developed through emulsion polymerization [73]. Administration of these nanoparticles induced significant anti-Tat IgG titers, with intramuscular vaccination promoting a Th1 immune response characterized by elevated IFN-γ and IL-2 responses, and reduced IL-4 levels [70]. The multifaceted potential of PMMA nanoparticles as vaccine adjuvants warrants further investigation in immunology.
4.2. Liposomes-based Vaccine Delivery
Liposomes are bilayer lipid vesicles composed of natural amphiphilic lipids and phospholipid molecules that offer diverse possibilities through the inclusion of components such as sterols, polypeptides, antioxidants, and polymers. These additional elements enable structural modulation, extended blood circulation, enhanced antioxidative properties, and targeted approaches to these lipid vesicles [74,75]. Liposomes improve the encapsulation, release, and delivery of bioactive compounds, thereby increasing their stability and effectiveness [75]. The preference for bilayered formulations with cholesterol and polyethylene glycol (PEG) incorporation optimizes cellular endocytosis and shields against immune cell attack [76].
The roots of liposomes in mRNA vaccine technology were traced back to 1978 when rabbit globin mRNA was delivered to mouse lymphocytes [76]. In recent decades, liposomes have been developed to enhance subunit vaccines [77]. However, optimizing liposome efficacy requires refinement of factors such as surface charge, size, and lipid bilayer composition [78]. Depending on the desired outcomes, a plethora of strategies enable the conjugation or encapsulation of ligands, such as drugs, peptides, cytokines, RNA or nucleotides, and antibodies within liposomes [79].
The drive to engineer liposomal vaccines stems from the goal of customizing immune responses by targeting specific immune cell subsets [80]. As delivery systems or adjuvants, cationic liposomes have the potential to augment various subunit vaccines, owing to their strong attraction to anionic immune cells [77]. Combining cationic liposomes with immunostimulatory factors enhances interactions with APCs, yielding robust cellular and humoral immune responses [81].
The utilization of virosome-based technology in approved liposomal vaccine formulations, such as Inflexal® V and Epaxal®, involves the coupling of viral proteins to the liposome surface [82]. Diverse techniques have been investigated to enhance formulation stability during storage [82], presenting promising avenues for advancing liposomal vaccines in future immunization strategies.
4.3. Micelle-based Vaccine Delivery
Micelles spontaneously undergo self-assembly in aqueous environments to form core-shell nanoparticles. The size and shape of these assemblies are dictated by thermodynamically driven self-assembly, which is influenced by hydrophilic and hydrophobic block sizes. Originally utilized for drug delivery by encapsulating hydrophobic compounds within the core, micellar nanoparticles have gained attention as promising vaccine delivery carriers [83].
4.3.1. PLA-based micelles
The utilization of PLA-based nanoparticles as adjuvants has attracted interest because of their favorable biodegradability and biocompatibility [84]. Jiménez-Sánchez et al. demonstrated the potential of micelles formed from a PLA-b-P(N-acryloxysuccinimide-co-N-vinylpyrrolidone) block copolymer [85]. These micelles allowed surface coupling of HIV-1 Gag p24 and encapsulation of imiquimod within the PLA core. Notably, encapsulated imiquimod demonstrated enhanced stimulation and maturation of DCs in vitro compared with its free form [85].
Jain et al. conducted a comparative assessment of the immunogenicity of hepatitis B surface antigen (HBsAg) using PLA polymer and PEG-PLA-PEG co-polymer formulations [86]. These findings demonstrated the superior efficacy of PEG-PLA-PEG micelles over PLA nanoparticles in augmenting and extending HBsAg-induced mucosal antibody responses following oral and intranasal immunization [87]. These findings underscore the potential of micelle-based nanosystems using a PLA platform for effective vaccine delivery.
4.3.2. Polypeptide-based micelles
Luo et al. (2013) pioneered the development of novel micelles using a PEG-b-poly(L-lysine) (PLL)-b-poly (L-leucine) architecture [88]. Through interactions between cationic PLL and anionic OVA and polyriboinosinic: Polyribocytidylic acid (PIC), a TLR3 agonist, these polypeptide micelles achieved simultaneous encapsulation of OVA and PIC, leading to synergistic enhancement of tumor-specific CTL responses. To address tumor-associated DC dysfunction linked to hyperactive STAT3 signaling, researchers incorporated STAT3 siRNA, PIC, and OVA within micelles for cancer vaccine purposes [89].
Investigating the adjuvant potential of γ-PGA micelles alongside influenza A viral antigen (PR8), researchers have noted significant outcomes. Intranasal PR8 immunization in the presence of the micelles led to elevated PR8-specific IgG levels in mouse sera and mucosal IgA antibody levels compared to PR8 immunization alone. Furthermore, PR8 with γ-PGA micelles induced robust IFN-γ-producing cells, indicating the capacity of the micelle system to serve as an effective delivery system eliciting both humoral and cellular immunity. Remarkably, mice immunized with PR8 and γ-PGA micelles demonstrated 100% immunity against the lethal PR8 virus [90]. This study highlights the potential of γ-PGA-based micelles in enhancing mucosal immunity and improving vaccine efficacy.
4.3.3. pH-responsive micelles
The use of pH-responsive micelles enhances the delivery of antigens to APCs in draining lymph nodes [91]. The subcutaneous injection of mice with OVA-polymer conjugates led to a significant increase in antigen-specific CD8+ T-cell responses, which was higher than the responses observed in mice immunized with soluble protein, OVA-polymer mixtures, or control micelle-immunized mice. Furthermore, the incorporation of a CpG ODN binding TLR9 into micelles amplified immune responses through electrostatic interactions with the cationic sections of the micelle [92].
Boudier et al. introduced pH-responsive micelles composed of polymethacrylic acid-b-polyethylene glycol/PLL for antigen peptide delivery [93]. In vitro investigations revealed the efficient loading, uptake, and release of antigen peptides in DCs. Furthermore, micelles notably induced DC maturation, underscoring their immunostimulatory properties [94]. This study emphasizes the potential of pH-responsive micelles to promote antigen uptake and DC activation, potentially enhancing the immune response.
4.4. Phage-based Vaccine Delivery
Bacteriophages (phages) hold significant potential as versatile subunit vaccine platforms owing to their favorable attributes such as size, surface architecture, safety, stability, biodegradability, and cost-effectiveness [95]. This section highlights various bacteriophages that offer distinct structural benefits for assembling and delivering pathogenic molecules, including proteins and DNAs, into VLP subunits, resulting in robust immune responses [Table 2].
Table 2: Bacteriophages used in virus-like particle-based subunit vaccine development [97].
| Parameters | M13 | T7 | λ | T4 | MS2 | Qβ |
|---|---|---|---|---|---|---|
| Capsid size (nm) | 900×9 | 56 | 60 | 120×86 | 26 | 28 |
| Phage protein (s) used for display (copies/capsid) | pVIII (2700) PIII (5) | gp10B (415) | gpD (405–420) | Hoc (155) Soc (870) | CP (180) | A1 (3–5) |
| Preferred molecule for in vivo display | Peptide | Peptide | Peptide | Full-length protein, Peptide | Peptide | Peptide |
| Maximum copy number | 2700 | 415 | 420 | 1025 | 180 | 86 |
| Main display scheme | In vivo | In vivo | In vivo In vitro | In vivo In vitro | In vivo | In vivo |
| Co-delivery of DNA (capacity) and protein | No | No | Possible (up to 48 kb) | Yes (up to 170 kb) | No | No |
| High density multiple antigen display | No | No | Possible (In vitro) | Yes | No | No |
| Targeted delivery of antigen | Yes | No | No | Yes | No | No |
| Adjustable copy number | No | No | Possible (In vitro) | Yes (In vitro) | No | No |
| Display of mammalian expressed antigen | No | No | Possible (In vitro) | Yes (In vitro) | No | No |
4.4.1. Phage T4
Utilizing insights from T4 structure and assembly, VLP subunit vaccines have been engineered against diverse pathogens, including Bacillus anthracis [96], Yersinia pestis [97], HIV-1 [98], foot-and-mouth disease virus (FMDV) [99], classical swine fever virus [100,101], and bursal disease virus [102].
A T4 VLP vaccine was engineered for anthrax by fusing protective antigens (PA) to the NH2-terminus of Hoc and assembling them on T4 capsids using hoc−soc−T4 phage nanoparticles [103]. Intramuscular administration of T4 displayed PA-induced 6.5-fold and 4.7-fold higher neutralizing antibodies against lethal toxins in mice, surpassing soluble PA immunization [103]. Moreover, Soc fusion variants of PA were also efficiently displayed on T4 phage through NH2-terminus or COOH-terminus fusion [104].
A T4 VLP-based vaccine for Y. pestis was also generated, resulting in approximately 660 F1mutV copies on each capsid [97]. Administered without an adjuvant, these T4 VLPs elicited robust F1V-specific antibodies, surpassing their soluble counterparts adjuvanted with Alhydrogel [105]. Notably, the T4 VLP-based vaccine generated a balanced Th1 and Th2 response, whereas the soluble F1mutV vaccine predominantly triggered Th2 and weak Th1 responses. This aligns with the potential of subunit vaccines to induce both innate and adaptive immunities. This was corroborated by the finding that the T4 VLP-based vaccine generated higher IFN-γ levels than the soluble F1mutV vaccine did. Critically, the T4 VLP-based vaccine conferred full protection against Y. pestis CO92 strain challenge even at high doses [105,106].
In addition to vaccines against bacterial pathogens, T4 phages have been used to develop vaccines against viral infections. Sathaliyawala et al. obtained HIV-1 p24-gag displayed on T4 capsids [98]. This yielded robust and durable anti-p24 antibody responses compared with the soluble p24 antigen, which generated weaker responses. Impressively, T4-p24 VLPs triggered potent CD4+ and CD8+ T-cell responses in contrast to soluble p24, which elicited limited responses [98].
4.4.2. Filamentous phages
Filamentous phages are extensively employed for presenting short random peptide libraries. Although these phages are modestly explored as vaccine-delivery systems to convey peptide antigens, their potential is slowly being recognized [107]. These elongated phages, approximately 900 nm in length, encompassed approximately 2700 copies of pVIII major capsid protein. Peptides derived from N20 pathogens have demonstrated substantial immunogenicity in diverse animal models, inducing robust cellular and humoral immunity [108,109]. Nevertheless, the assembly of filamentous phages necessitates extrusion of the pVIII capsid protein, making the display size-dependent. Typically, short peptides containing B- or T-cell epitopes are ideal for antigen presentation [110]. Although larger peptides can be displayed, their copy numbers are constrained because of their incorporation alongside the wild-type capsid protein [111]. Although both pIII and pVIII capsid proteins are deployable, the limited copy number of pIII diminishes their attractiveness for vaccine delivery.
4.4.3. Phage λ
The icosahedral capsid of the λ phage, measuring 60 nm, formed both the hexagonal capsid lattice and the majority of pentameric vertices. In addition, 405–420 copies of gpD embellish the capsid, adopting trimeric arrangements on quasi-three-fold axes [112]. In contrast to T4 phage Soc, gpD plays an essential role in stabilizing the capsid enclosing the 48.5 kb genome [113], although its necessity diminishes for capsids carrying shorter genomes [114]. They have been widely applied in peptide displays [115]. Although both amino- and carboxy-termini are suitable for antigen peptide fusion [116], the apparent interaction of the amino-terminus with gpE makes it a less preferred choice [117]. Therefore, the carboxy-terminus is the preferred site for displaying antigenic peptides and proteins [118,119].
4.4.4. Phage T7
The T7 capsid, measuring 56 nm and enclosing a 40 kb genome, contains two capsid proteins: gp10A and gp10B [120]. Although gp10B arises from a −1 frameshift at the COOH-terminus of the gp10A reading frame, this is not crucial for phage capsid assembly. Consequently, gp10B is harnessed in phage display, allowing antigenic peptides to fuse at the COOH terminus [121]. This strategy effectively displayed antigenic peptides with up to 50 amino acids. Notably, Tan et al. established strong immunogenicity of a 46-amino acid HBsAg peptide conjugated to T7 phages in rabbits [122]. In addition, Xu et al. demonstrated that a T7 phage with a 40-aa GH loop peptide of FMDV VP1 exhibited high immunogenicity and conferred 80% survivability after the swine virus challenge [123]. Similarly, displaying the ectodomain of the influenza virus channel protein M2 (24 amino acids) on T7 elicited robust cellular and humoral responses, effectively safeguarding mice against challenges from influenza H1N1 and H3N2 viruses [124].
4.4.5. Phage MS2
By employing the “two-domain” approach, Peabody et al. effectively demonstrated the high immunogenicity of an MS2 phage by displaying a 10aa residue from the V3 loop peptide of the HIV envelope [125]. Similarly, the MS2 phage presenting a 15aa peptide epitope from the minor capsid protein of HPV16 generates neutralizing antibodies, conferring protection against different HPV pseudovirus types in vivo [126]. However, certain peptide insertions can affect capsid protein assembly. Basu et al. found that 5 out of 6 Zika virus envelope protein B-cell epitopes disrupted MS2 VLP assembly, hampering CP assembly [127]. Heal et al. also demonstrated that MS2 phage presenting the malaria parasite Plasmodium falciparum’s protective epitope T1 elicited robust immunity in vivo [128]. Dong et al. highlighted the high immunogenicity of phage MS2 with FMDV epitopes in vivo, which induced significant levels of neutralizing antibodies [129]. Although these instances underscore the effectiveness of MS2 VLPs for delivering short antigens, it is evident that they are less suitable for conveying larger antigens.
4.4.6. Phage Qβ
Qβ, a compact bacteriophage, has been utilized for antigen delivery [130]. The Qβ phage capsid is 28 nm in diameter and comprises 180 copies of the major coat protein [131]. Antigenic proteins and peptides can be presented on the capsid through fusion with the read-through domain of A1 [132]. Optimization by Vasiljeva et al. shortened the read-through domain to only 6aa, boosting A1 copy numbers to 86 per capsid [133]. On the other hand, antigen peptides can be linked to the Qβ capsid [134]. Qβ phage-displaying antigens exhibit remarkable immunogenicity [135]. Intranasal administration of Qβ VLPs showcasing the M2 protein ectodomain of the influenza virus triggered robust M2-specific IgG and IgA production in a mouse model, safeguarding against influenza virus challenge [135]. Furthermore, Qβ has been explored for generating vaccine candidates against non-infectious ailments such as hypertension, nicotine addiction, diabetes, cancer, Alzheimer’s disease, and allergies, with six candidates advancing to Phase I or II clinical trials [136].
4.5. Hydrogel-based Vaccine Delivery
Various studies have examined polymeric hydrogel-based vaccine delivery platforms owing to their capacity and effectiveness in antigen delivery. Hydrogel systems possess distinctive attributes, efficiently directing antigens/vaccines to specific anatomical/physiological sites, potentially serving as delivery systems, and aiding antigen-triggered immune responses [137].
Peptide-based gels and nanogels have emerged as effective platforms for the delivery of vaccines. Li et al. introduced a novel peptide nanofiber hydrogel serving as a carrier for respiratory syndrome virus vaccines [138]. Supramolecular peptide hydrogels have been developed as carriers for West Nile virus vaccines to induce significant immune responses [139]. Peptide-hydrogel systems are promising vaccine adjuvants, robustly enhancing antigen-triggered immune responses [140]. A nanogel for nasal vaccine delivery was formulated, whereas another nanogel-based system effectively delivered immunogenic proteins through interactions with ethylenediamine groups, demonstrating high antigen delivery efficiency [141]. Notably, nanogel devices efficiently entrap and interact with antigenic molecules, utilizing hydrophobic interactions within the polymeric gel network [142].
Various injectable hydrogel-based systems were developed for effective vaccine delivery. Wu et al. designed an injectable hydrogel from PCL and PEG, exhibiting notable immunogenicity upon antigen exposure [143]. Another injectable hydrogel harnessed a pentablock copolymer of PEG, PLA, and PCL for sustained vaccine release and demonstrated significant antigen-specific immunity [144]. A novel injectable self-assembled hydrogel composed of poly (L-valine) was developed for the dual delivery of antigens and TLR agonists, which exhibited prolonged antigen persistence and antitumor effects in melanoma-bearing mice [145]. In addition, an injectable hydrogel formulation was engineered to encapsulate both the model antigen (OVA) and granulocyte-macrophage colony-stimulating factor, effectively delivering antigens and enhancing immunogenicity [146]. A vaccine system combining PEG-b-poly (L-alanine) facilitated the co-delivery of an immune checkpoint inhibitor and tumor vaccine, successfully inducing tumor-infiltrating CD8+ T cells and was effective against B16F10 melanoma [147]. An injectable polypeptide hydrogel was introduced for sustained cargo antigen delivery [148]. Furthermore, injectable polymer-nanoparticle hydrogels have been developed for the efficient delivery of subunit vaccines, expanding the array of vaccine delivery strategies [149].
4.6. Inorganic-based Vaccine Delivery
Researchers have extensively explored the application of inorganic NPs in vaccine development. These NPs, characterized by their controllable synthesis and rigid structure, offer advantages for vaccine delivery. However, their limited biodegradability is a concern. Inorganic NPs were employed as carriers and adjuvants to augment immune responses. Notable examples of inorganic NPs include silica, carbon, aluminum-based, calcium phosphate, magnetic, and gold nanoparticles [150].
Gold nanoparticles (AuNPs) can be readily shaped (spherical, rod-shaped, cubic, etc.), and their diverse forms can elicit both cellular and humoral responses [151]. By attaching antigens to gold nanorods, they effectively delivered respiratory syncytial virus antigens [152]. Other forms of AuNPs have been used as adjuvants for DNA vaccines against HIV and as carriers for antigens from viruses, such as influenza and foot-and-mouth disease [153,154]. Carbon NPs were also engineered into mesoporous spheres and nanotubes and linked to protein and peptide antigens to amplify the IgG response [155]. Silica-based NPs are good nanocarriers for vaccine delivery, targeting specific tumors [156], and enabling real-time multimodal imaging [157]. Structural adjustments enable these nanoparticles to selectively interact with cells [158]. Calcium phosphate nanoparticles represent another class of inorganic nanoparticles formed by combining sodium citrate, dibasic sodium phosphate, and calcium chloride under specific conditions [159]. These non-toxic nanoparticles can range in size from 50 nm to 100 nm [160].
Gold nanorods were functionalized with polyethyleneimine, resulting in remarkable enhancement of humoral and cellular immunity. This effect is attributed to the activation of APCs. This improvement was observed in comparison with treatment with naked HIV envelope plasmid DNA in vivo [161]. Wang et al. conducted research involving the conjugation of trimetric influenza HA to AuNPs. They also employed the TLR-5 agonist flagellin as an adjuvant. This approach triggers the proliferation of CD4+ and CD8+ cells upon intranasal vaccination in mice, subsequently elevating influenza-specific IgA and IgG antibody titers [162]. In addition, investigations have indicated that certain types of inorganic NPs can induce toxic effects in the male reproductive system of rodents [163].
4.7. Emulsion-based Vaccine Delivery
Emulsions showed a significant part in vaccine formulation and are currently being investigated for their potential use in vaccine delivery systems. Due to their inherent thermodynamic instability, emulsions can segregate into distinct oil and water phases [164]. They have been employed to administer vaccines by incorporating antigens into their structures or combining them with antigens. Nanoemulsions demonstrate better performance in delivering antigens to APCs than larger emulsions because of their ability to effectively penetrate the nasal mucosa. Many of these nanoemulsions are used as adjuvants during vaccine formulation [165].
Microemulsions (MEs) represent a novel class of vaccine delivery systems with enhanced target specificity and therapeutic efficacy compared to nanoemulsions owing to their spontaneous generation and thermodynamic stability [166]. Researchers have shown that MEs can enhance the immune-boosting effects of flavonoid compounds and adjuvants used in influenza vaccines when administered nasally. In addition, ME formulations comprising propylene glycol, isopropyl myristate, and polysorbate 80 as carriers for rabies and bluetongue virus vaccines have yielded no topical reactions [167]. MEs proved highly effective in rabies immunization while showing limited humoral immunity for the bluetongue vaccine, possibly due to particle size-mediated adjuvanticity control. Particle sizes of 20–50 nm facilitate optimal cellular absorption, promoting enhanced uptake into the lymphatic system and DC activation [168]. Emulsifiers, such as Cremophor (CreEL, Polyoxyl 35 castor oil) and Solutol HS15 (Macrogol 15 hydroxystearate), which mitigate interfacial tension and confer emulsion stability, are crucial for the spontaneous generation of effective MEs [169].
To combat Acinetobacter baumannii infections, Yang et al. created a vaccine that combined the OmpK/Omp22 fusion protein with MF59, a vaccine adjuvant composed of an oil-in-water emulsion containing squalene and two surfactants, polysorbate 80 and sorbitan trioleate [170]. These constituents were emulsified within citrate buffer, yielding droplets of approximately 160 nm in diameter. Following intratracheal immunization and two booster doses in BALB/c mice, this approach yielded neutralizing antibodies, diminished bacterial concentrations in lung and blood tissues, and abated inflammatory cytokines [170].
5. CONCLUSIONS
In this comprehensive review, we explore an array of subunit vaccine delivery systems, each offering unique attributes to enhance immunogenicity and therapeutic efficacy. The diversity of these vaccine delivery systems highlights the dynamic landscape of vaccine development driven by the pursuit of safer and more effective vaccination strategies. Through the synthesis of research findings, we delineated the capabilities and limitations of polymer-based, lipid-based, micelle-based, phage-based, hydrogel-based, inorganic-based, and emulsion-based platforms.
Despite significant progress in subunit vaccine delivery, several challenges remain. There remains a need to decipher the complex interplay between the physicochemical properties of carriers and immune response outcomes. In addition, the long-term safety and biocompatibility of these systems warrant further scrutiny, particularly in the context of human applications. Comparative studies elucidating the relative strengths of different systems and their compatibility with diverse antigens are essential.
The future holds promising direction for subunit vaccine delivery. Integrating cutting-edge technologies, such as nanomedicine, gene editing, and synthetic biology, could unleash new frontiers for enhanced antigen presentation and immune modulation. Rational design approaches based on structural biology and computational modeling will foster the creation of precisely engineered carriers. Tailoring delivery systems to specific target populations, such as the elderly or immunocompromised, could enhance the efficacy of vaccines. Advances in personalized medicine may enable the customization of vaccines based on individual immune profiles.
In conclusion, the rapid evolution of subunit vaccine delivery systems has marked an exciting era in vaccinology. As we navigate the intricate landscape of immune responses and harness the power of innovative delivery platforms, we are poised to shape a future in which vaccines are safer, more effective, and accessible to all.
6. ACKNOWLEDGMENTS
The authors would like to thank the Department of Science and Technology (DOST) Fellows Program, the Philippine Council for Agriculture, Aquatic and Natural Resources Research and Development (DOST-PCAARRD) for funding this research project, and the Industrial Technology Development Institute for hosting this research project.
7. AUTHOR CONTRIBUTIONS
All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; agreed to submit to the current journal; gave final approval of the version to be published.
8. FUNDING
This project was funded by the DOST-PCAARRD.
9. CONFLICTS OF INTEREST
The authors report no financial or any other conflicts of interest in this work.
10. ETHICAL APPROVALS
This study does not involve experiments on animals or human subjects.
11. DATA AVAILABILITY
All the data is available with the authors and shall be provided upon request.
12. USE OF ARTIFICIAL INTELLIGENCE (AI)-ASSISTED TECHNOLOGY
The authors declares that they have not used artificial intelligence (AI)-tools for writing and editing of the manuscript, and no images were manipulated using AI.
12. PUBLISHER’S NOTE
All claims expressed in this article are solely those of the authors and do not necessarily represent those of the publisher, the editors and the reviewers. This journal remains neutral with regard to jurisdictional claims in published institutional affiliation.
REFERENCES
1. Rappuoli R, Pizza M, Del Giudice G, De Gregorio E. Vaccines, new opportunities for a new society. Proc Natl Acad Sci U S A 2014;111:12288-93. [CrossRef]
2. Tognotti E. The eradication of smallpox, a success story for modern medicine and public health:What lessons for the future?J Infect Dev Ctries 2010;4:264-6.
3. Finco O, Rappuoli R. Designing vaccines for the twenty-first century society. Front Immunol 2014;5:12. [CrossRef]
4. Delany I, Rappuoli R, De Gregorio E. Vaccines for the 21st century. EMBO Mol Med 2014;6:708-20. [CrossRef]
5. Mahedi MR, Rawat A, Rabbi F, Babu KS, Tasayco ES, Areche FO, et al. Understanding the global transmission and demographic distribution of Nipah Virus (NiV). Res J Pharm Technol 2023;16:3588-94. [CrossRef]
6. Yadav DK, Yadav N, Khurana SM. Vaccines:Present status and applications. In:Verma AS, Singh A, editors. Animal Biotechnology. 2nd ed., Ch. 26. Boston:Academic Press;2020. 523-42.
7. Karch CP, Burkhard P. Vaccine technologies:From whole organisms to rationally designed protein assemblies. Biochem Pharmacol 2016;120:1-14. [CrossRef]
8. Pollard AJ, Bijker EM. A guide to vaccinology:From basic principles to new developments. Nat Rev Immunol 2021;21:83-100. [CrossRef]
9. Zhang Y, Chen Y, Zhou J, Wang X, Ma L, Li J, et al. Porcine epidemic diarrhea virus:An updated overview of virus epidemiology, virulence variation patterns and virus-host interactions. Viruses 2022;14:2434. [CrossRef]
10. Brito LA, O'Hagan DT. Designing and building the next generation of improved vaccine adjuvants. J Control Release 2014;190:563-79. [CrossRef]
11. Levitz SM, Golenbock DT. Beyond empiricism:Informing vaccine development through innate immunity research. Cell 2012;148:1284-92. [CrossRef]
12. Lee KL, Twyman RM, Fiering S, Steinmetz NF. Virus-based nanoparticles as platform technologies for modern vaccines. Wiley Interdiscip Rev Nanomed Nanobiotechnol 2016;8:554-78. [CrossRef]
13. Mooney M, McWeeney S, Canderan G, Sékaly RP. A systems framework for vaccine design. Curr Opin Immunol 2013;25:551-5. [CrossRef]
14. Odendall C, Kagan JC. Activation and pathogenic manipulation of the sensors of the innate immune system. Microbes Infect 2017;19:229-37. [CrossRef]
15. Schenten D, Medzhitov R. The control of adaptive immune responses by the innate immune system. Adv Immunol 2011;109:87-124. [CrossRef]
16. Steinman RM, Banchereau J. Taking dendritic cells into medicine. Nature 2007;449:419-26. [CrossRef]
17. Liu Z, Qiu D, Wang F, Taylor JA, Zhang M. Grain refinement of cast zinc through magnesium inoculation:Characterisation and mechanism. Mater Charact 2015;106:1-10. [CrossRef]
18. Austyn JM. Dendritic cells in the immune system-history, lineages, tissues, tolerance, and immunity. In:Myeloid Cells in Health and Disease. Hoboken:John Wiley and Sons, Ltd.;2017. 155-207.
19. Lutz MB, Schuler G. Immature, semi-mature and fully mature dendritic cells:Which signals induce tolerance or immunity?Trends Immunol 2002;23:445-9.
20. Delaloye J, Roger T, Steiner-Tardivel QG, Le Roy D, Knaup Reymond M, Akira S, et al. Innate immune sensing of modified Vaccinia virus Ankara (MVA) is mediated by TLR2-TLR6, MDA-5 and the NALP3 inflammasome. PLoS Pathog 2009;5:e1000480.
21. Pulendran B, Ahmed R. Immunological mechanisms of vaccination. Nat Immunol 2011;12:509-17. [CrossRef]
22. Banchereau J, Steinman RM. Dendritic cells and the control of immunity. Nature 1998;392:245-52. [CrossRef]
23. Janeway CA Jr., Travers P, Walport M, Shlomchik MJ. Immunobiology. 5th ed. New York:Garland Science;2001.
24. Zhang N, Bevan MJ. CD8(+) T cells:Foot soldiers of the immune system. Immunity 2011;35:161-8. [CrossRef]
25. Blum JS, Wearsch PA, Cresswell P. Pathways of antigen processing. Annu Rev Immunol 2013;31:443-73. [CrossRef]
26. Fehres CM, Unger WW, Garcia-Vallejo JJ, van Kooyk Y. Understanding the biology of antigen cross-presentation for the design of vaccines against cancer. Front Immunol 2014;5:149. [CrossRef]
27. Zhu J, Yamane H, Paul WE. Differentiation of effector CD4 T cell populations (*). Annu Rev Immunol 2010;28:445-89. [CrossRef]
28. Luckheeram RV, Zhou R, Verma AD, Xia B. CD4+T cells:Differentiation and functions. Clin Dev Immunol 2012;2012:925135. [CrossRef]
29. Harwood NE, Batista FD. Early events in B cell activation. Annu Rev Immunol 2010;28:185-210. [CrossRef]
30. Nutt SL, Hodgkin PD, Tarlinton DM, Corcoran LM. The generation of antibody-secreting plasma cells. Nat Rev Immunol 2015;15:160-71. [CrossRef]
31. MuŽíkováG, Laga R. Macromolecular systems for vaccine delivery. Physiol Res 2016;65:S203-16.
32. Cubas R, Zhang S, Kwon S, Sevick-Muraca EM, Li M, Chen C, et al. Virus-like particle (VLP) lymphatic trafficking and immune response generation after immunization by different routes. J Immunother 2009;32:118-28. [CrossRef]
33. Azad N, Rojanasakul Y. Vaccine delivery--current trends and future. Curr Drug Deliv 2006;3:137-46. [CrossRef]
34. Johansen P, Kündig TM. Parenteral vaccine administration:Tried and true. In:Foged C, Rades T, Perrie Y, Hook S, editors. Subunit Vaccine Delivery. Advances in Delivery Science and Technology. New York:Springer;2015. 261-86.
35. Herzog C. Influence of parenteral administration routes and additional factors on vaccine safety and immunogenicity:A review of recent literature. Expert Rev Vaccines 2014;13:399-415. [CrossRef]
36. Lambert PH, Laurent PE. Intradermal vaccine delivery:Will new delivery systems transform vaccine administration?Vaccine 2008;26:3197-208.
37. Hunter P, Fryhofer SA, Szilagyi PG. Vaccination of adults in general medical practice. Mayo Clin Proc 2020;95:169-83. [CrossRef]
38. Lemoine C, Thakur A, Krajišnik D, Guyon R, Longet S, Razim A, et al. Technological approaches for improving vaccination compliance and coverage. Vaccines (Basel) 2020;8:304. [CrossRef]
39. Bacon A, Makin J, Sizer PJ, Jabbal-Gill I, Hinchcliffe M, Illum L, et al. Carbohydrate biopolymers enhance antibody responses to mucosally delivered vaccine antigens. Infect Immun 2000;68:5764-70. [CrossRef]
40. Nevagi RJ, Skwarczynski M, Toth I. Polymers for subunit vaccine delivery. Eur Polym J 2019;114:397-410. [CrossRef]
41. Sarmento B, das Neves J. Chitosan-based Systems for Biopharmaceuticals:Delivery, Targeting and Polymer Therapeutics. Hoboken:John Wiley and Sons;2012. 691.
42. Zhang J, Xia W, Liu P, Cheng Q, Tahirou T, Gu W, et al. Chitosan modification and pharmaceutical/biomedical applications. Mar Drugs 2010;8:1962-87. [CrossRef]
43. Zaharoff DA, Rogers CJ, Hance KW, Schlom J, Greiner JW. Chitosan solution enhances both humoral and cell-mediated immune responses to subcutaneous vaccination. Vaccine 2007;25:2085-94. [CrossRef]
44. Li X, Min M, Du N, Gu Y, Hode T, Naylor M, et al. Chitin, chitosan, and glycated chitosan regulate immune responses:The novel adjuvants for cancer vaccine. Clin Dev Immunol 2013;2013:e387023.
45. Marasini N, Skwarczynski M, Toth I. Intranasal delivery of nanoparticle-based vaccines. Ther Deliv 2017;8:151-67. [CrossRef]
46. Jain S, Khomane K, Jain AK, Dani P. Nanocarriers for transmucosal vaccine delivery. Curr Nanosci 2011;7:160-77. [CrossRef]
47. Sui Z, Chen Q, Fang F, Zheng M, Chen Z. Cross-protection against influenza virus infection by intranasal administration of M1-based vaccine with chitosan as an adjuvant. Vaccine 2010;28:7690-8. [CrossRef]
48. Otterlei M, Ostgaard K, Skjåk-Braek G, Smidsrød O, Soon-Shiong P, Espevik T. Induction of cytokine production from human monocytes stimulated with alginate. J Immunother (1991) 1991;10:286-91. [CrossRef]
49. Farjaha A, Owlia P, Siadat SD, Mousavi SF, Shafieeardestani M. Conjugation of alginate to a synthetic peptide containing T-and B-cell epitopes as an induction for protective immunity against Pseudomonas aeruginosa. J Biotechnol 2014;192:240-7. [CrossRef]
50. Kogan G, Soltés L, Stern R, Gemeiner P. Hyaluronic acid:A natural biopolymer with a broad range of biomedical and industrial applications. Biotechnol Lett 2007;29:17-25. [CrossRef]
51. Necas J, Bartosikova L, Brauner P, Kolar J. Hyaluronic acid (hyaluronan):A review. Vet Med 2008;53:397-411. [CrossRef]
52. Kong WH, Sung DK, Kim H, Yang JA, Ieronimakis N, Kim KS, et al. Self-adjuvanted hyaluronate--antigenic peptide conjugate for transdermal treatment of muscular dystrophy. Biomaterials 2016;81:93-103. [CrossRef]
53. Termeer C, Benedix F, Sleeman J, Fieber C, Voith U, Ahrens T, et al. Oligosaccharides of hyaluronan activate dendritic cells via toll-like receptor 4. J Exp Med 2002;195:99-111. [CrossRef]
54. Gariboldi S, Palazzo M, Zanobbio L, Selleri S, Sommariva M, Sfondrini L, et al. Low molecular weight hyaluronic acid increases the self-defense of skin epithelium by induction of beta-defensin 2 via TLR2 and TLR4. J Immunol 2008;181:2103-10. [CrossRef]
55. Lees A, Finkelman F, Inman JK, Witherspoon K, Johnson P, Kennedy J, et al. Enhanced immunogenicity of protein-dextran conjugates:I. Rapid stimulation of enhanced antibody responses to poorly immunogenic molecules. Vaccine 1994;12:1160-6. [CrossRef]
56. Shinchi H, Crain B, Yao S, Chan M, Zhang SS, Ahmadiiveli A, et al. Enhancement of the immunostimulatory activity of a TLR7 ligand by conjugation to polysaccharides. Bioconjug Chem 2015;26:1713-23. [CrossRef]
57. Zhang W, An M, Xi J, Liu H. Targeting CpG adjuvant to lymph node via dextran conjugate enhances antitumor immunotherapy. Bioconjug Chem 2017;28:1993-2000. [CrossRef]
58. Luo M, Shao B, Nie W, Wei XW, Li YL, Wang BL, et al. Antitumor and adjuvant activity of λ-carrageenan by stimulating immune response in cancer immunotherapy. Sci Rep 2015;5:11062. [CrossRef]
59. Zhang YQ, Tsai YC, Monie A, Hung CF, Wu TC. Carrageenan as an adjuvant to enhance peptide-based vaccine potency. Vaccine 2010;28:5212-9. [CrossRef]
60. Sinha VR, Bansal K, Kaushik R, Kumria R, Trehan A. Poly-epsilon-caprolactone microspheres and nanospheres:An overview. Int J Pharm 2004;278:1-23. [CrossRef] [CrossRef]
61. Jameela SR, Suma N, Misra A, Raghuvanshi R, Ganga S, Jayakrishnan A. Poly(ε-caprolactone) microspheres as a vaccine carrier. Curr Sci 1996;70:669-71.
62. Slobbe L, Medlicott N, Lockhart E, Davies N, Tucker I, Razzak M, et al. Aprolonged immune response to antigen delivered in poly (epsilon-caprolactone) microparticles. Immunol Cell Biol 2003;81:185-91. [CrossRef]
63. Cruz LJ, Tacken PJ, Eich C, Rueda F, Torensma R, Figdor CG. Controlled release of antigen and toll-like receptor ligands from PLGA nanoparticles enhances immunogenicity. Nanomedicine (Lond) 2017;12:491-510. [CrossRef]
64. Hamdy S, Haddadi A, Hung RW, Lavasanifar A. Targeting dendritic cells with nano-particulate PLGA cancer vaccine formulations. Adv Drug Deliv Rev 2011;63:943-55. [CrossRef]
65. Bailey BA, Desai KH, Ochyl LJ, Ciotti SM, Moon JJ, Schwendeman SP. Self-encapsulating poly(lactic-co-glycolic acid) (PLGA) microspheres for intranasal vaccine delivery. Mol Pharm 2017;14:3228-37. [CrossRef]
66. Inbaraj BS, Chen BH. In vitro removal of toxic heavy metals by poly(γ-glutamic acid)-coated superparamagnetic nanoparticles. Int J Nanomedicine 2012;7:4419-32.
67. Matsusaki M, Hiwatari Ken-Ichiro, Higashi M, Kaneko T, Akashi M. Stably-dispersed and surface-functional bionanoparticles prepared by self-assembling amphipathic polymers of hydrophilic poly(γ-glutamic acid) bearing hydrophobic amino acids. Chem Lett 2004;33:398-9. [CrossRef]
68. Uto T, Wang X, Sato K, Haraguchi M, Akagi T, Akashi M, et al. Targeting of antigen to dendritic cells with poly(gamma-glutamic acid) nanoparticles induces antigen-specific humoral and cellular immunity. J Immunol 2007;178:2979-86. [CrossRef]
69. Akagi T, Wang X, Uto T, Baba M, Akashi M. Protein direct delivery to dendritic cells using nanoparticles based on amphiphilic poly(amino acid) derivatives. Biomaterials 2007;28:3427-36. [CrossRef]
70. Bettencourt A, Almeida AJ. Poly(methyl methacrylate) particulate carriers in drug delivery. J Microencapsul 2012;29:353-67. [CrossRef]
71. Kreuter J, Speiser PP. New adjuvants on a polymethylmethacrylate base. Infect Immun 1976;13:204-10. [CrossRef]
72. Eldridge JH, Hammond CJ, Meulbroek JA, Staas JK, Gilley RM, Tice TR. Controlled vaccine release in the gut-associated lymphoid tissues. I. Orally administered biodegradable microspheres target the Peyer's patches. J Control Release 1990;11:205-14. [CrossRef]
73. Voltan R, Castaldello A, Brocca-Cofano E, Altavilla G, Caputo A, Laus M, et al. Preparation and characterization of innovative protein-coated poly(methylmethacrylate) core-shell nanoparticles for vaccine purposes. Pharm Res 2007;24:1870-82. [CrossRef]
74. Bolhassani A. Lipid-based delivery systems in development of genetic and subunit vaccines. Mol Biotechnol 2023;65:669-98. [CrossRef]
75. Raoufi E, Bahramimeimandi B, Salehi-Shadkami M, Chaosri P, Mozafari MR. Methodical design of viral vaccines based on avant-garde nanocarriers:A multi-domain narrative review. Biomedicines 2021;9:520. [CrossRef]
76. Okay S, Özcan ÖÖ, Karahan M, Okay S, Özcan ÖÖ, Karahan M. Nanoparticle-based delivery platforms for mRNA vaccine development. AIMS Biophys 2020;7:323-38. [CrossRef]
77. Tretiakova DS, Vodovozova EL. Liposomes as adjuvants and vaccine delivery systems. Biochem (Mosc) Suppl Ser A Membr Cell Biol 2022;16:1-20.
78. Luwi NE, Ahmad S, Azlyna AS, Nordin A, Sarmiento ME, Acosta A, et al. Liposomes as immunological adjuvants and delivery systems in the development of tuberculosis vaccine:A review. Asian Pac J Trop Med 2022;15:7. [CrossRef]
79. Fobian SF, Cheng Z, Ten Hagen TL. Smart lipid-based nanosystems for therapeutic immune induction against cancers:Perspectives and outlooks. Pharmaceutics 2021;14:26. [CrossRef]
80. De Serrano LO, Burkhart DJ. Liposomal vaccine formulations as prophylactic agents:Design considerations for modern vaccines. J Nanobiotechnology 2017;15:83. [CrossRef]
81. Khademi F, Taheri RA, Momtazi-Borojeni AA, Farnoosh G, Johnston TP, Sahebkar A. Potential of cationic liposomes as adjuvants/delivery systems for tuberculosis subunit vaccines. Rev Physiol Biochem Pharmacol 2018;175:47-69. [CrossRef]
82. Tang J, Cai L, Xu C, Sun S, Liu Y, Rosenecker J, et al. Nanotechnologies in delivery of DNA and mRNA vaccines to the nasal and pulmonary mucosa. Nanomaterials (Basel) 2022;12:226. [CrossRef]
83. Croy SR, Kwon GS. Polymeric micelles for drug delivery. Curr Pharm Des 2006;12:4669-84. [CrossRef]
84. Pavot V, Rochereau N, Primard C, Genin C, Perouzel E, Lioux T, et al. Encapsulation of Nod1 and Nod2 receptor ligands into poly(lactic acid) nanoparticles potentiates their immune properties. J Control Release 2013;167:60-7. [CrossRef]
85. Jiménez-Sánchez G, Pavot V, Chane-Haong C, HandkéN, Terrat C, Gigmes D, et al. Preparation and in vitro evaluation of imiquimod loaded polylactide-based micelles as potential vaccine adjuvants. Pharm Res 2015;32:311-20. [CrossRef]
86. Jain AK, Goyal AK, Gupta PN, Khatri K, Mishra N, Mehta A, et al. Synthesis, characterization and evaluation of novel triblock copolymer based nanoparticles for vaccine delivery against hepatitis B. J Control Release 2009;136:161-9. [CrossRef]
87. Jain AK, Goyal AK, Mishra N, Vaidya B, Mangal S, Vyas SP. PEG-PLA-PEG block copolymeric nanoparticles for oral immunization against hepatitis B. Int J Pharm 2010;387:253-62. [CrossRef]
88. Luo Z, Li P, Deng J, Gao N, Zhang Y, Pan H, et al. Cationic polypeptide micelle-based antigen delivery system:A simple and robust adjuvant to improve vaccine efficacy. J Control Release 2013;170:259-67. [CrossRef]
89. Luo Z, Wang C, Yi H, Li P, Pan H, Liu L, et al. Nanovaccine loaded with poly I:C and STAT3 siRNA robustly elicits anti-tumor immune responses through modulating tumor-associated dendritic cells in vivo. Biomaterials 2015;38:50-60. [CrossRef]
90. Noh YW, Hong JH, Shim SM, Park HS, Bae HH, Ryu EK, et al. Polymer nanomicelles for efficient mucus delivery and antigen-specific high mucosal immunity. Angew Chem Int Ed Engl 2013;52:7684-9. [CrossRef]
91. Keller S, Wilson JT, Patilea GI, Kern HB, Convertine AJ, Stayton PS. Neutral polymer micelle carriers with pH-responsive, endosome-releasing activity modulate antigen trafficking to enhance CD8(+) T cell responses. J Control Release 2014;191:24-33. [CrossRef]
92. Wilson JT, Keller S, Manganiello MJ, Cheng C, Lee CC, Opara C, et al. pH-responsive nanoparticle vaccines for dual-delivery of antigens and immunostimulatory oligonucleotides. ACS Nano 2013;7:3912-25. [CrossRef]
93. Boudier A, Aubert-Pouëssel A, Louis-Plence P, Gérardin C, Jorgensen C, Devoisselle JM, et al. The control of dendritic cell maturation by pH-sensitive polyion complex micelles. Biomaterials 2009;30:233-41. [CrossRef]
94. Boudier A, Aubert-Pouëssel A, Mebarek N, Chavanieu A, Quentin J, Martire D, et al. Development of tripartite polyion micelles for efficient peptide delivery into dendritic cells without altering their plasticity. J Control Release 2011;154:156-63. [CrossRef]
95. Fu Y, Li J. A novel delivery platform based on bacteriophage MS2 virus-like particles. Virus Res 2016;211:9-16. [CrossRef]
96. Peachman KK, Li Q, Matyas GR, Shivachandra SB, Lovchik J, Lyons RC, et al. Anthrax vaccine antigen-adjuvant formulations completely protect New Zealand white rabbits against challenge with Bacillus anthracis Ames strain spores. Clin Vaccine Immunol 2012;19:11-6. [CrossRef]
97. Tao P, Mahalingam M, Rao VB. Highly effective soluble and bacteriophage T4 nanoparticle plague vaccines against Yersinia pestis. Methods Mol Biol 2016;1403:499-518. [CrossRef]
98. Sathaliyawala T, Rao M, Maclean DM, Birx DL, Alving CR, Rao VB. Assembly of human immunodeficiency virus (HIV) antigens on bacteriophage T4:A novel in vitro approach to construct multicomponent HIV vaccines. J Virol 2006;80:7688-98. [CrossRef]
99. Ren ZJ, Tian CJ, Zhu QS, Zhao MY, Xin AG, Nie WX, et al. Orally delivered foot-and-mouth disease virus capsid protomer vaccine displayed on T4 bacteriophage surface:100% protection from potency challenge in mice. Vaccine 2008;26:1471-81. [CrossRef]
100. Orosco FL. Current progress in diagnostics, therapeutics, and vaccines for African swine fever virus. Vet Integr Sci 2023;21:751-81. [CrossRef]
101. Wu J, Tu C, Yu X, Zhang M, Zhang N, Zhao M, et al. Bacteriophage T4 nanoparticle capsid surface SOC and HOC bipartite display with enhanced classical swine fever virus immunogenicity:A powerful immunological approach. J Virol Methods 2007;139:50-60. [CrossRef]
102. Cao YC, Shi QC, Ma JY, Xie QM, Bi YZ. Vaccination against very virulent infectious bursal disease virus using recombinant T4 bacteriophage displaying viral protein VP2. Acta Biochim Biophys Sin (Shanghai) 2005;37:657-64. [CrossRef]
103. Shivachandra SB, Rao M, Janosi L, Sathaliyawala T, Matyas GR, Alving CR, et al. In vitro binding of anthrax protective antigen on bacteriophage T4 capsid surface through Hoc-capsid interactions:A strategy for efficient display of large full-length proteins. Virology 2006;345:190-8. [CrossRef]
104. Li Q, Shivachandra SB, Zhang Z, Rao VB. Assembly of the small outer capsid protein, Soc, on bacteriophage T4:A novel system for high density display of multiple large anthrax toxins and foreign proteins on phage capsid. J Mol Biol 2007;370:1006-19. [CrossRef]
105. Tao P, Mahalingam M, Kirtley ML, van Lier CJ, Sha J, Yeager LA, et al. Mutated and bacteriophage T4 nanoparticle arrayed F1-V immunogens from Yersinia pestis as next generation plague vaccines. PLoS Pathog 2013;9:e1003495.
106. Vela Ramirez JE, Sharpe LA, Peppas NA. Current state and challenges in developing oral vaccines. Adv Drug Deliv Rev 2017;114:116-31. [CrossRef]
107. Henry KA, Arbabi-Ghahroudi M, Scott JK. Beyond phage display:Non-traditional applications of the filamentous bacteriophage as a vaccine carrier, therapeutic biologic, and bioconjugation scaffold. Front Microbiol 2015;6:755. [CrossRef]
108. Deng L, Roose K, Job ER, De Rycke R, Van Hamme E, Gonçalves A, et al. Oral delivery of Escherichia coli persistently infected with M2e-displaying bacteriophages partially protects against influenza A virus. J Control Release 2017;264:55-65. [CrossRef]
109. Orosco FL. Immune evasion mechanisms of porcine epidemic diarrhea virus:A comprehensive review. Vet Integr Sci 2024;22:171-92. [CrossRef]
110. Prisco A, De Berardinis P. Filamentous bacteriophage fd as an antigen delivery system in vaccination. Int J Mol Sci 2012;13:5179-94. [CrossRef]
111. Samoylova TI, Braden TD, Spencer JA, Bartol FF. Immunocontraception:Filamentous bacteriophage as a platform for vaccine development. Curr Med Chem 2017;24:3907-20. [CrossRef]
112. Nicastro J, Sheldon K, Slavcev RA. Bacteriophage lambda display systems:Developments and applications. Appl Microbiol Biotechnol 2014;98:2853-66. [CrossRef]
113. Hernando-Pérez M, Lambert S, Nakatani-Webster E, Catalano CE, de Pablo PJ. Cementing proteins provide extra mechanical stabilization to viral cages. Nat Commun 2014;5:4520. [CrossRef]
114. Sternberg N, Hoess RH. Display of peptides and proteins on the surface of bacteriophage lambda. Proc Natl Acad Sci U S A 1995;92:1609-13. [CrossRef]
115. Catalano CE. Bacteriophage lambda:The path from biology to theranostic agent. Wiley Interdiscip Rev Nanomed Nanobiotechnol 2018;10:e1517.
116. Chang JR, Song EH, Nakatani-Webster E, Monkkonen L, Ratner DM, Catalano CE. Phage lambda capsids as tunable display nanoparticles. Biomacromolecules 2014;15:4410-9. [CrossRef]
117. Lander GC, Evilevitch A, Jeembaeva M, Potter CS, Carragher B, Johnson JE. Bacteriophage lambda stabilization by auxiliary protein gpD:Timing, location, and mechanism of attachment determined by cryo-EM. Structure 2008;16:1399-406. [CrossRef]
118. González-Cano P, Gamage LN, Marciniuk K, Hayes C, Napper S, Hayes S, et al. Lambda display phage as a mucosal vaccine delivery vehicle for peptide antigens. Vaccine 2017;35:7256-63. [CrossRef]
119. Gamage LN, Ellis J, Hayes S. Immunogenicity of bacteriophage lambda particles displaying porcine circovirus 2 (PCV2) capsid protein epitopes. Vaccine 2009;27:6595-604. [CrossRef]
120. Guo F, Liu Z, Fang PA, Zhang Q, Wright ET, Wu W, et al. Capsid expansion mechanism of bacteriophage T7 revealed by multistate atomic models derived from cryo-EM reconstructions. Proc Natl Acad Sci U S A 2014;111:E4606-14.
121. Xu GJ, Kula T, Xu Q, Li MZ, Vernon SD, Ndung'u T, et al. Viral immunology. Comprehensive serological profiling of human populations using a synthetic human virome. Science 2015;348:aaa0698.
122. Tan GH, Yusoff K, Seow HF, Tan WS. Antigenicity and immunogenicity of the immunodominant region of hepatitis B surface antigen displayed on bacteriophage T7. J Med Virol 2005;77:475-80. [CrossRef]
123. Xu H, Bao X, Lu Y, Liu Y, Deng B, Wang Y, et al. Immunogenicity of T7 bacteriophage nanoparticles displaying G-H loop of foot-and-mouth disease virus (FMDV). Vet Microbiol 2017;205:46-52. [CrossRef]
124. Hashemi H, Pouyanfard S, Bandehpour M, Noroozbabaei Z, Kazemi B, Saelens X, et al. Immunization with M2e-displaying T7 bacteriophage nanoparticles protects against influenza A virus challenge. PLoS One 2012;7:e45765.
125. Peabody DS, Manifold-Wheeler B, Medford A, Jordan SK, do Carmo Caldeira J, Chackerian B. Immunogenic display of diverse peptides on virus-like particles of RNA phage MS2. J Mol Biol 2008;380:252-63. [CrossRef]
126. Tumban E, Peabody J, Tyler M, Peabody DS, Chackerian B. VLPs displaying a single L2 epitope induce broadly cross-neutralizing antibodies against human papillomavirus. PLoS One 2012;7:e49751.
127. Basu R, Zhai L, Contreras A, Tumban E. Immunization with phage virus-like particles displaying Zika virus potential B-cell epitopes neutralizes Zika virus infection of monkey kidney cells. Vaccine 2018;36:1256-64. [CrossRef]
128. Heal KG, Hill HR, Stockley PG, Hollingdale MR, Taylor-Robinson AW. Expression and immunogenicity of a liver stage malaria epitope presented as a foreign peptide on the surface of RNA-free MS2 bacteriophage capsids. Vaccine 1999;18:251-8. [CrossRef]
129. Dong YM, Zhang GG, Huang XJ, Chen L, Chen HT. Promising MS2 mediated virus-like particle vaccine against foot-and-mouth disease. Antiviral Res 2015;117:39-43. [CrossRef]
130. Pumpens P, Renhofa R, Dishlers A, Kozlovska T, Ose V, Pushko P, et al. The true story and advantages of RNA phage capsids as nanotools. Intervirology 2016;59:74-110. [CrossRef]
131. Rumnieks J, Tars K. Crystal structure of the maturation protein from bacteriophage Qβ. J Mol Biol 2017;429:688-96. [CrossRef]
132. Brown SD, Fiedler JD, Finn MG. Assembly of hybrid bacteriophage Qbeta virus-like particles. Biochemistry 2009;48:11155-7. [CrossRef]
133. Vasiljeva I, Kozlovska T, Cielens I, Strelnikova A, Kazaks A, Ose V, et al. Mosaic Qbeta coats as a new presentation model. FEBS Lett 1998;431:7-11. [CrossRef]
134. Kündig TM, Senti G, Schnetzler G, Wolf C, Prinz Vavricka BM, Fulurija A, et al. Der p 1 peptide on virus-like particles is safe and highly immunogenic in healthy adults. J Allergy Clin Immunol 2006;117:1470-6. [CrossRef]
135. Bessa J, Schmitz N, Hinton HJ, Schwarz K, Jegerlehner A, Bachmann MF. Efficient induction of mucosal and systemic immune responses by virus-like particles administered intranasally:Implications for vaccine design. Eur J Immunol 2008;38:114-26. [CrossRef]
136. Huang X, Wang X, Zhang J, Xia N, Zhao Q. Escherichia coli-derived virus-like particles in vaccine development. NPJ Vaccines 2017;2:3. [CrossRef]
137. Roth GA, Gale EC, Alcántara-Hernández M, Luo W, Axpe E, Verma R, et al. Injectable hydrogels for sustained codelivery of subunit vaccines enhance humoral immunity. ACS Cent Sci 2020;6:1800-12. [CrossRef]
138. Li X, Galliher-Beckley A, Huang H, Sun X, Shi J. Peptide nanofiber hydrogel adjuvanted live virus vaccine enhances cross-protective immunity to porcine reproductive and respiratory syndrome virus. Vaccine 2013;31:4508-15. [CrossRef]
139. Friedrich BM, Beasley DW, Rudra JS. Supramolecular peptide hydrogel adjuvanted subunit vaccine elicits protective antibody responses against West Nile virus. Vaccine 2016;34:5479-82. [CrossRef]
140. Wang Y, Zhang J, Wang Y, Wang K, Wei H, Shen L. Isolation and characterization of the Bacillus cereus BC7 strain, which is capable of zearalenone removal and intestinal flora modulation in mice. Toxicon 2018;155:9-20. [CrossRef]
141. Zhang T, Yang R, Yang S, Guan J, Zhang D, Ma Y, et al. Research progress of self-assembled nanogel and hybrid hydrogel systems based on pullulan derivatives. Drug Deliv 2018;25:278-92. [CrossRef]
142. Salatin S, Barar J, Barzegar-Jalali M, Adibkia K, Milani MA, Jelvehgari M. Hydrogel nanoparticles and nanocomposites for nasal drug/vaccine delivery. Arch Pharm Res 2016;39:1181-92. [CrossRef]
143. Wu Q, Gong C, Shi S, Wang Y, Huang M, Yang L, et al. Mannan loaded biodegradable and injectable thermosensitive PCL-PEG-PCL hydrogel for vaccine delivery. Soft Mater 2012;10:472-86. [CrossRef]
144. Bobbala S, Tamboli V, McDowell A, Mitra AK, Hook S. Novel injectable pentablock copolymer based thermoresponsive hydrogels for sustained release vaccines. AAPS J 2016;18:261-9. [CrossRef]
145. Song H, Huang P, Niu J, Shi G, Zhang C, Kong D, et al. Injectable polypeptide hydrogel for dual-delivery of antigen and TLR3 agonist to modulate dendritic cells in vivo and enhance potent cytotoxic T-lymphocyte response against melanoma. Biomaterials 2018;159:119-29. [CrossRef]
146. Sun Z, Liang J, Dong X, Wang C, Kong D, Lv F. Injectable hydrogels coencapsulating granulocyte-macrophage colony-stimulating factor and ovalbumin nanoparticles to enhance antigen uptake efficiency. ACS Appl Mater Interfaces 2018;10:20315-25. [CrossRef]
147. Song H, Yang P, Huang P, Zhang C, Kong D, Wang W. Injectable polypeptide hydrogel-based co-delivery of vaccine and immune checkpoint inhibitors improves tumor immunotherapy. Theranostics 2019;9:2299-314. [CrossRef]
148. Asai D, Fukuda T, Morokuma K, Funamoto D, Yamaguchi Y, Mori T, et al. Injectable polypeptide hydrogel depot system for assessment of the immune response-inducing efficacy of sustained antigen release alone. Macromol Biosci 2019;19:e1900167.
149. Zhang L, Yang W, Hu C, Wang Q, Wu Y. Properties and applications of nanoparticle/microparticle conveyors with adjuvant characteristics suitable for oral vaccination. Int J Nanomedicine 2018;13:2973-87. [CrossRef]
150. Bezbaruah R, Chavda VP, Nongrang L, Alom S, Deka K, Kalita T, et al. Nanoparticle-based delivery systems for vaccines. Vaccines (Basel) 2022;10:1946. [CrossRef]
151. Niikura K, Matsunaga T, Suzuki T, Kobayashi S, Yamaguchi H, Orba Y, et al. Gold nanoparticles as a vaccine platform:Influence of size and shape on immunological responses in vitro and in vivo. ACS Nano 2013;7:3926-38. [CrossRef]
152. Stone JW, Thornburg NJ, Blum DL, Kuhn SJ, Wright DW, Crowe JE Jr. Gold nanorod vaccine for respiratory syncytial virus. Nanotechnology 2013;24:295102. [CrossRef]
153. Tao W, Ziemer KS, Gill HS. Gold nanoparticle-M2e conjugate coformulated with CpG induces protective immunity against influenza A virus. Nanomedicine (Lond) 2014;9:237-51. [CrossRef]
154. Chen YS, Hung YC, Lin WH, Huang GS. Assessment of gold nanoparticles as a size-dependent vaccine carrier for enhancing the antibody response against synthetic foot-and-mouth disease virus peptide. Nanotechnology 2010;21:195101. [CrossRef]
155. Wang T, Zou M, Jiang H, Ji Z, Gao P, Cheng G. Synthesis of a novel kind of carbon nanoparticle with large mesopores and macropores and its application as an oral vaccine adjuvant. Eur J Pharm Sci 2011;44:653-9. [CrossRef]
156. Ingle SG, Pai RV, Monpara JD, Vavia PR. Liposils:An effective strategy for stabilizing paclitaxel loaded liposomes by surface coating with silica. Eur J Pharm Sci 2018;122:51-63. [CrossRef]
157. Benezra M, Penate-Medina O, Zanzonico PB, Schaer D, Ow H, Burns A, et al. Multimodal silica nanoparticles are effective cancer-targeted probes in a model of human melanoma. J Clin Invest 2011;121:2768-80. [CrossRef]
158. Niut Y, Popatt A, Yu M, Karmakar S, Gu W, Yu C. Recent advances in the rational design of silica-based nanoparticles for gene therapy. Ther Deliv 2012;3:1217-37. [CrossRef]
159. He Q, Mitchell AR, Johnson SL, Wagner-Bartak C, Morcol T, Bell SJ. Calcium phosphate nanoparticle adjuvant. Clin Diagn Lab Immunol 2000;7:899-903. [CrossRef]
160. Joyappa DH, Kumar CA, Banumathi N, Reddy GR, Suryanarayana VV. Calcium phosphate nanoparticle prepared with foot and mouth disease virus P1-3CD gene construct protects mice and guinea pigs against the challenge virus. Vet Microbiol 2009;139:58-66. [CrossRef]
161. Xu L, Liu Y, Chen Z, Li W, Liu Y, Wang L, et al. Surface-engineered gold nanorods:Promising DNA vaccine adjuvant for HIV-1 treatment. Nano Lett 2012;12:2003-12. [CrossRef]
162. Wang C, Zhu W, Luo Y, Wang BZ. Gold nanoparticles conjugating recombinant influenza hemagglutinin trimers and flagellin enhanced mucosal cellular immunity. Nanomedicine 2018;14:1349-60. [CrossRef]
163. Dantas GP, Ferraz FS, Andrade LM, Costa GM. Male reproductive toxicity of inorganic nanoparticles in rodent models:A systematic review. Chem Biol Interact 2022;363:110023. [CrossRef]
164. Chavda VP, Shah D. Self-emulsifying delivery systems:One step ahead in improving solubility of poorly soluble drugs. In:Ficai A, Grumezescu AM, editors. Nanostructures for Cancer Therapy. Micro and Nano Technologies. Ch. 25. Amsterdam:Elsevier;2017. 653-718.
165. O'Hagan DT. MF59 is a safe and potent vaccine adjuvant that enhances protection against influenza virus infection. Expert Rev Vaccines 2007;6:699-710. [CrossRef]
166. Nasiri MI, Vora LK, Ershaid JA, Peng K, Tekko IA, Donnelly RF. Nanoemulsion-based dissolving microneedle arrays for enhanced intradermal and transdermal delivery. Drug Deliv Transl Res 2022;12:881-96. [CrossRef]
167. Lee JJ, Shim A, Lee SY, Kwon BE, Kim SR, Ko HJ, et al. Ready-to-use colloidal adjuvant systems for intranasal immunization. J Colloid Interface Sci 2016;467:121-8. [CrossRef]
168. Jiang W, Kim BY, Rutka JT, Chan WC. Nanoparticle-mediated cellular response is size-dependent. Nat Nanotechnol 2008;3:145-50. [CrossRef]
169. Lamaisakul S, Tantituvanont A, Lipipun V, Ritthidej G. Development of novel cationic microemulsion as parenteral adjuvant for influenza vaccine. Asian J Pharm Sci 2020;15:591-604. [CrossRef]
170. Yang AQ, Yang HY, Guo SJ, Xie YE. MF59 adjuvant enhances the immunogenicity and protective immunity of the OmpK/Omp22 fusion protein from Acineterbacter baumannii through intratracheal inoculation in mice. Scand J Immunol 2019;90:e12769.