
1. INTRODUCTION
Since the emergence of coronavirus disease (COVID-19) in late 2019, SARS-CoV-2 has undergone continuous evolution through the accumulation of mutations in its genome. Being an RNA virus, the absence of a repair system results in a high mutation rate for SARS-CoV-2 [1]. Consequently, the world records a very high genetic diversity, with thousands of variants of SARS-CoV-2 already circulating. Some variants have emerged to possess more infectious and virulent mutations. Variants with a high degree of health threat, high infectious rate, and significant immune evasion have been identified as variants of concern (VOC). There have been rising and retrogressing VOC variants Alpha, Beta, Gamma, and Delta [2].
Omicron is a variant classified as VOC (PANGO Lineages designation B.1.1.529) that was first recorded in Botswana and South Africa in November 2021 [3]. Omicron is rapidly spreading to all six continents and was classified as a VOC by the WHO on November 26, 2021 [4]. Studies have shown that Omicron has a faster transmission rate than all previously recorded VOCs [5-7]. As a result, Omicron replaced Delta globally and became the main circulating variant [8]. Genetic analysis indicates that the original Omicron is significantly different across the genome than previous VOC variants. Therefore, a clear conclusion about the origin of Omicron has yet to be reached [9]. Genetic analysis has also shown that Omicron’s Spike (S) gene has the highest number of mutations, totaling 35, which result in 30 amino acid substitutions [10]. The mutations appearing in the S gene are believed to be the primary reason for the substantial expansion of Omicron. The region with the most mutations (15 mutations) is the receptor-binding domain (RBD), which encodes a motif that directly binds to the hACE2 receptor. Mutations in this domain potentially confer an evolutionary advantage by enhancing viral ACE2-RBD binding or avoiding neutralizing antibodies [11]. It has not stopped here, and Omicron continued to infect and generate novel subvariants with better characterizations. At present, five groups of Omicron subvariants are circulating globally, namely, BA.1*, BA.2*, BA.3*, BA.4*, BA.5*, and Omicron subvariants resulting from recombination (combination variants – CBV) [12]. The genetic and classified differences among these six Omicron subvariants are based on variations within the S gene [13,14]. Therefore, continuing to evaluate and understand the genetic diversity and evolutionary direction of the S gene will provide more information for molecular research of Omicron.
Vietnam is known as one of the few countries that managed to control COVID-19 outbreaks from the end-2019 to mid-2021 [15]. It was only with the appearance of the Delta variant that the government imposed restrictions and stringent defensive measures, which were accompanied by a rapid increase in infections and deaths. Similar to the COVID-19 situation worldwide, by the end of 2021, Vietnam recorded the arrival of the Omicron variant. Subsequently, Omicron became the primary variant causing infections in Hanoi in March 2022. In a previous study in May 2022, we showed that Omicron appeared in Vietnam, including two variants, BA.1* and BA.2*, in which BA.2* dominated. There have yet to be any published molecular investigations of Omicron, although this variant continues introducing many subvariants into Vietnam. Hence, the objective of this study is to offer the most recent information regarding the prevalence and genetic diversity of the Omicron variant isolated in Vietnam, based on its S gene sequence.
2. MATERIALS AND METHODS
2.1. Data Collection
The whole genome and subsequent information of SARS-CoV-2 Omicron variants isolated in Vietnam were collected from GISAID; the reference S gene of Wuhan-hu-1 sequence was downloaded in GenBank (accession number: NC_045512.2) [16]. To extract the S gene of Omicron variants, the whole genomes were aligned with the reference sequence by MAFFT server [17]. The aligned sequences were used to find and extract the S gene from whole genome in MEGA software [18].
2.2. Haplotype/Identification and Mutation Scanning
Mutations appeared in the S gene were investigated by an in-house Perl script. Consequently, all the S gene sequences were grouped, and assigned haplotype number by DnaSP 6.12.03 [19] then was used for further analysis.
2.3. Phylogenetics and Haplotype Network Analysis
A maximum likelihood (ML) phylogenetic tree was constructed using a bootstrap resampling approach with a value of 1000 using IQtree 2.2.0 [20]. The GTR+F+I+G4 nucleotide substitution model was used as the best model for building ML tree based on the BIC score of IQtree. The resulting tree topology provides insights into the evolutionary connections and branching patterns of the entities within the dataset, with branch support values reflecting the degree of confidence in each inferred relationship. The Minimum Spanning Network of Omicron haplotypes was built using infomation generated by PopART [21]. Gephi software was used to visualize and calculate the centrality of each node in the network.
2.4. Genetic Diversity and Molecular Distances Calculating
The genetic diversity measures (haplotype and nucleotide diversity) and molecular distance analysis of Omicron and its subvariants were determined by Arlequin 3.5 [22]. The t-test in R packages was used to find out significantly statistic analysis.
3. RESULTS
3.1. The Haplotype Diversity
There were a total of 6348 sequences of Omicron collected from Vietnam and reported in the GISAID database from December 2021 to December 2023. In which, BA.2* variants were the most common, accounting for 52.13% of the sequences. BA.5* variants were the second most common, accounting for 28.94% of the sequences. BA.1* variants accounted for 6.30% of the sequences, and XBB* variants accounted for 11.91% of the sequences [Table 1]. The Omicron subvariant had a high level of haplotype diversity, which implies high genetic diversity [Table 1]. The haplotype analysis for each subvariant showed that BA.2* had the highest number of haplotypes, followed by BA.5*, XBB*, BA.1*, and BA.4* [Table 1]. This diversity within BA.2* and BA.5* correlates with their global prevalence [23,24], highlighting the influence of globally circulating subvariants on Omicron’s dynamics in Vietnam.
Table 1: The diversity of Omicron subvariants appeared in Vietnam.
| Frequency (%) | Number of haplotypes | Haplotype diversity | |
|---|---|---|---|
| BA.1* | 6.30 | 119 | 0.9409±0.0060 |
| BA.2* | 52.13 | 934 | 0.9236±0.0033 |
| BA.4* | 0.49 | 21 | 0.9591±0.0214 |
| BA.5* | 28.94 | 482 | 0.8720±0.0061 |
| XBB* | 11.91 | 292 | 0.9453±0.0057 |
Although XBB* has just appeared in Vietnam in early 2023, but it had a higher number of haplotypes and diversity when compared to BA.1* and BA.4*, suggesting that XBB* could be the potential to become the predominant circulating variant in Vietnam afterward. In addition to XBB*, Vietnam has also recorded the presence of other Omicron CBVs, such as XBL (with four sequences), XAZ (with three sequences), and XBF (with three sequences). The emergence and cocirculation of these Omicron subvariants in Vietnam from late 2022 to early 2023 closely resemble the global epidemiological patterns observed.
3.2. The Nucleotide Diversity
Our further analysis of the S gene haplotypes of Omicron in Vietnam revealed 1090 polymorphic sites, encompassing 762 transitions and 353 transversions. In which, 178 insertion/deletion (indel) sites were identified specific to the NTD encoding region. Figure 1a provides a visual representation of the locations and frequencies of polymorphic sites introduced in the S gene. Notably, there are significant clusters of mutations, particularly in S1, and the RBD region exhibits a noteworthy preservation of missense mutations [Figure 1a]. In addition, the analysis indicated a prominent prevalence of missense mutations throughout the S gene, suggesting a positive selection adaptation of this gene [Figure 1b].
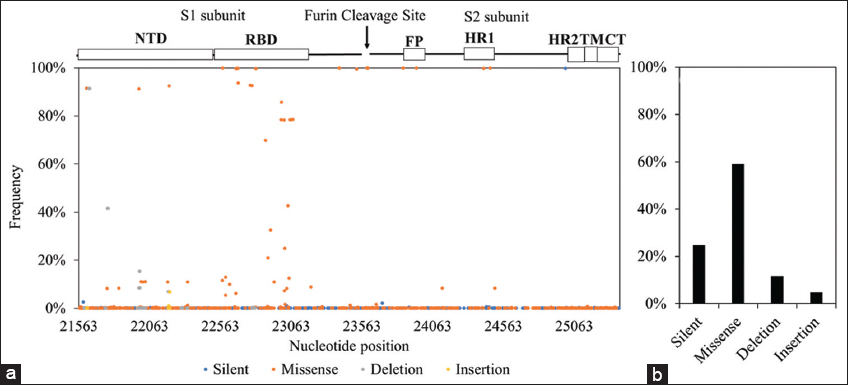 | Figure 1: The distribution and examination of mutation in the S gene. (a) Position mutations in the S gene. Upper part of the diagram shows the relative positions of the subunits S1, S2 (and their sub-regions) on the S gene. The Y-axis of chart is mutation frequency, and the X-axis is nucleotide position in S gene. The color, including blue, orange, gray, and yellow dots in chart, displays silent, missense, deletion, and insertion mutations, respectively. (b) The frequency of mutations appearing in S gene of Omicron variants isolated in Vietnam. The Y-axis displays frequency, and the X-axis is a types of mutation, including silent, missense, deletion, and insertion). [Click here to view] |
Examining the mutation that appeared in the S gene of Omicron, indicating an individual availability average of 42.9 ± 7.6 mutation [Figure 2a]. Mutation distribution is not equal to two subunit regions, in which the region encoded S1 subunit (S1) had higher mutation numbers at 34.5 ± 7.3 when compared to the S2 subunit (S2) at 8.4 ± 0.7 (P < 0.0001) [Figure 2a]. In addition, in S1, we found that the NTD region had 16.8 ± 4.7 mutations, higher than the RBD region, at 16.5 ± 4.6 (P < 0.006) [Figure 2a].
 | Figure 2: The mutations of the S gene of Omicron and subvariants. (a) The amount of mutation in the S gene of Omicron individual, including the compositions that encoded S1 subunit (NTD and RBD) and S2 subunit. The t-test was used to identify the differences in mutation in each region, with P < 0.05 displayed for the statistical significance. (b) The nucleotide spectrum in the S gene of Omicron variants. (c) The nucleotide changes and polymorphic sites of Omicron subvariants. (d) The amount of mutation of each Omicron subvariants. All error bars in this figure show the ± SD: standard deviation). [Click here to view] |
The analysis of mutations in each Omicron subvariant revealed variations in the mutation count among subvariants [Figure 2d]. Specifically, BA.1* and XBB* displayed the highest mutation counts, with 54.7 ± 5.9 and 54.2 ± 2.0 mutations, respectively, followed by BA.4 with 42.5 ± 4.0 and BA.5* with 41.7 ± 4.9 mutations [Figure 2d]. In contrast, the BA.2* variant exhibited the lowest mutation count among other subvariants, at 39.5 ± 5.7. Overall, the mutation among these Omicron subvariants converged in that the S1 region displayed concentrated mutations, while the S2 region remained highly conserved [Figure 2d]. BA.2* and XBB* exhibited a more significant number of RBD mutations than NTD, a habit not observed in other variants such as BA.1*, BA.4*, and BA.5* [Figure 2d]. These findings align with previous research, which noted that BA.2* had a higher mutation count in the RBD compared to BA.1*[25]. In the RBD region, notable highly conserved missense mutations include G22578A (G339D), T22679C (S373P), C22686T (S375F), G22813T (K417N), T22882G (N440K), G22992A (S477N), C22995A (T478K), A23013C (E484A), A23055G (Q498R), A23063T (N501Y), and T23075C (Y505H), all of which contribute to enhanced transmission of the Omicron variant [26,27].
Nucleotide change analysis revealed a predominance of transition mutations, particularly C to T substitutions, and reversals in Omicron [Figure 2b]. Among transitions, the G to T substitution exhibited the highest frequency. Notably, the frequency of G to T substitution has declined in Omicron variants worldwide [28]. In Vietnam, the G to T substitution frequency of 11.11% remains lower than the average observed in other variants such as Alpha and Delta (16.8%–18.5%) and previous publications [29,30]. Nucleotide diversity analysis indicated a similarity in nucleotide substitution patterns among Omicron subvariants, characterized by a high rate of transitions and a low rate of transversions [Figure 2b and c]. BA.2* and BA.5* stood out with significantly higher polymorphic sites than other subvariants [Figure 2c]. The elevated number of polymorphic sites in BA.2* and BA.5* correlates with our previous analyses.
Nucleotide diversity analysis showed that BA.1* exhibited the highest nucleotide diversity and nucleotide differences [Table 2]. This increased nucleotide diversity in BA.1* could attributed to indel mutations, primarily localized in the NTD region. Subsequently, the elevated diversity and nucleotide differences observed in BA.2* and BA.5* are consistent with the prolonged prevalence of these variants in Vietnam [Table 1]. In contrast, the lower diversity in XBB* corresponds to its recent emergence and circulation compared to the previous variants. However, overall, the nucleotide diversity indices of Omicron subvariants are higher than those of the variants previously reported in Vietnam, such as Alpha and Delta [29,30].
Table 2: The S gene nucleotide diversity of Omicron subvariants.
| Variants | Nucleotide diversity | Nucleotide pairwise difference |
|---|---|---|
| BA.1* | 0.003057±0.001535 | 11.712870±5.315446 |
| BA.2* | 0.002789±0.001404 | 10.742860±4.891336 |
| BA.4* | 0.002191±0.001160 | 8.339785±3.969329 |
| BA.5* | 0.001722±0.000900 | 6.603859±3.120736 |
| XBB* | 0.001133±0.000621 | 4.322580±2.141502 |
3.3. Molecular Distances of Omicron Subvariants
The maximum-likelihood (ML) tree was constructed to elucidate the relationships among Omicron subvariants based on their S gene sequences [Figure 3]. The ML analysis revealed that BA.2* and BA.5* displayed the most intricate patterns of differentiation. BA.2* exhibited substantial genetic diversity, evident from its multiple clusters within the ML tree. Similarly, BA.5* demonstrated high diversity, dividing into two prominent clusters. Furthermore, BA.1* was closely related to BA.2*, with a bootstrap value of 78.4. Concurrently, the XBB* variant formed a distinct clade and exhibited a close genetic relationship with the BA.2* group, supported by a bootstrap value of 97.3.
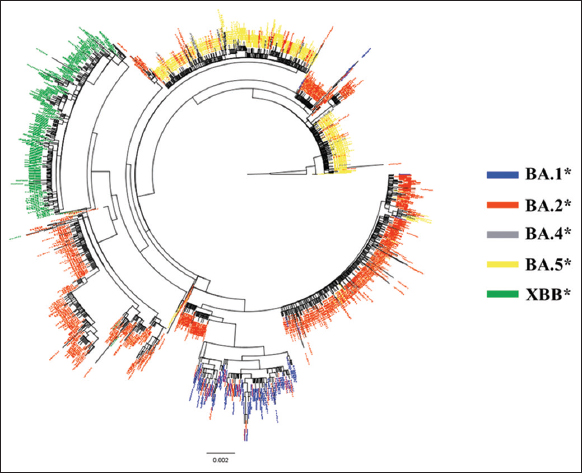 | Figure 3: The maximum-likelihood phylogenetic tree of the Omicron’s S gene. (The maximum-likelihood tree was built by IQtree software with 1000 bootstrap values. Each color of variant on this tree (blue, orange, black, yellow, and green presentation for BA.1*, BA.2*, BA.4*, BA.5*, and XBB*, respectively). [Click here to view] |
To ascertain the genetic relationships among Omicron subvariants, molecular distance analysis employing the AMOVA algorithm within Arlequin was conducted [Figure 4]. The results of molecular distances were based on two key parameters: nucleotide differences (DA) and genetic distance (FST). The AMOVA analysis indicated that BA.1* exhibited complete genetic independence from other variants, as evidenced by both and values. In contrast, BA.2* exhibited a notably close genetic affinity with BA.4*, BA.5*, and XBB*, with XBB* demonstrating the closest genetic relationship to BA.2*. This was evident through at 16.15725 nucleotides and ( value of 0.62824 (P < 0.01) when comparing with other Omicron subvariants. The close relationship between BA.2* and XBB* aligns with previous whole genome analyses, supporting the accuracy of our study [13,31,32]. In addition, a significant genetic association between BA.2* and BA.4* emerged, as indicated by (DA) and (FST) values of 7.59309 nucleotide differences and 0.41438, respectively. Similarly, a genetic kinship between BA.2* and BA.5* was observed, with (DA) and (FST) values of 6.93667 nucleotides and 0.42817, respectively. The genetic connection between BA.4* and BA.5* was illuminated (DA) and (FST) values of 0.77507 nucleotides and 0.10808 (P < 0.01) in comparison with other Omicron subvariants. Notably, BA.4* and BA.5* were initially identified in Gauteng in December 2021, approximately 1 month following the detection of BA.2* in the same region [33]. This temporal and geographical alignment substantiates the findings of our molecular distance analyses.
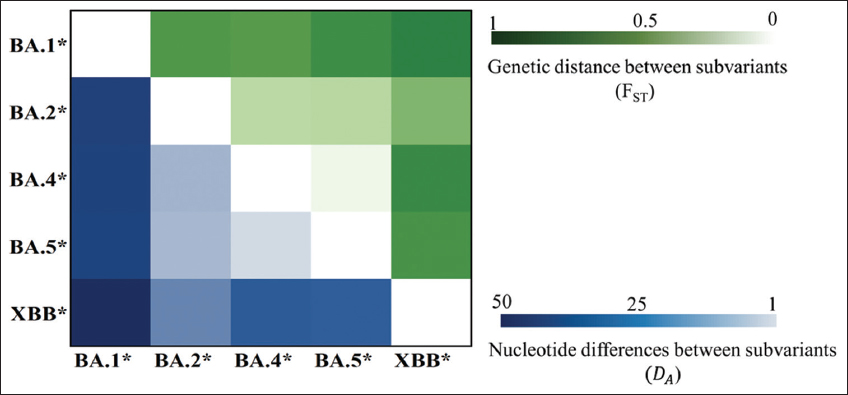 | Figure 4: The molecular distance between Omicron subvariants. (Molecular distances among Omicron subvariants using two metrics: nucleotide differences (DA), represented by a blue scale, and genetic distance (FST), depicted in green. A lighter color scale indicates a closer genetical, while a darker hue signifies greater genetic divergence). [Click here to view] |
The genetic relationships among Omicron subvariants and their roles in transmission and lineage emergence were assessed using centrality analysis of haplotype networks within the Gephi software. This analysis relied on two crucial measures: Betweenness Centrality (BC) and Eigenvector Centrality (EC). BC identifies haplotypes that act as critical haplotype connectors between other haplotypes in a network, playing a pivotal role in maintaining network cohesion. Conversely, EC identifies haplotypes with the most significant influence on the entire network based on their effectiveness in propagating through the network, determined by assessing extended connections. The BC analysis highlighted that the most influential haplotypes in the emergence of Omicron in Vietnam were associated with BA.2* [Figure 5a]. This observation corresponds with the central role of BA.2* identified in the molecular distances analysis [Figure 4]. Combining BC with EC revealed that all three haplotypes of BA.2* displayed high BC and EC values [Figure 5b]. This suggests that these three haplotypes directly contributed to generating offspring haplotypes and other variants. These findings indicate that these three haplotypes likely play important roles in facilitating high-efficiency transmission within the BA.2* lineage in Vietnam. Predictions based on BC and EC analyses strongly support the vision that BA.2* exhibits the highest predicted linkage efficiency compared to other Omicron variants, such as BA.1*, BA.4*, and BA.5* [34].
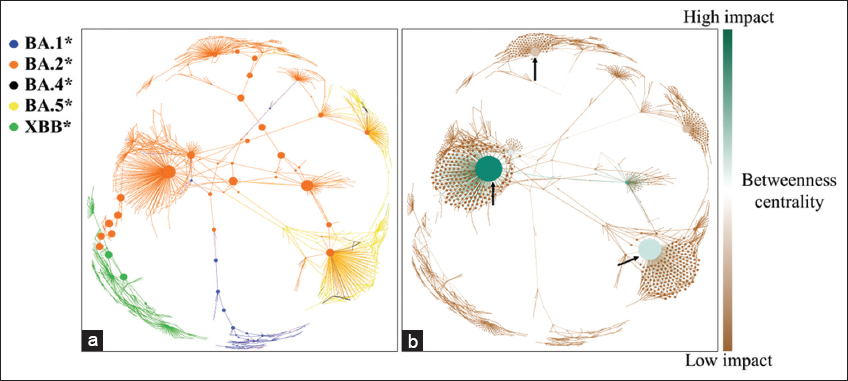 | Figure 5: The S gene haplotype network analysis of Omicron subvariant in Vietnam. (The S gene haplotype network of Omicron subvariants, with each haplotype represented as a circular node within the network. (a) Different colors represent each haplotype belonging to the Omicron subvariants, and the size of the nodes represents the betweenness centrality (BC) – larger nodes indicate higher BC values. (b) The combination network of both BC and eigenvector centrality (EC), where the size of each circular node reflects the EC value, and the color indicates the level of BC). [Click here to view] |
4. DISCUSSION
The comprehensive analysis of Omicron S gene sequences from Vietnam revealed important insights into the prevalence and diversity of various subvariants in the region. The study analyzed a total of 6348 sequences from the GISAID database, providing a robust dataset for understanding the dynamics of Omicron in Vietnam. The most common variant observed was BA.2*, accounting for 52.13% of the sequences, followed by BA.5* at 28.94%. These findings align with global prevalence data [23,24], indicating that globally circulating subvariants have a significant impact on the dynamics of Omicron in Vietnam. The high frequency of BA.2* and BA.5* suggests their sustained transmission and potential adaptation to the local population.
Haplotype analysis revealed a high level of genetic diversity within the Omicron subvariant in Vietnam. BA.2* exhibited the highest number of haplotypes, followed by BA.5*, XBB*, BA.1*, and BA.4*. This diversity within BA.2* and BA.5* further supports their global prevalence and underscores their impact on local transmission dynamics. Notably, XBB*, which emerged recently in early 2023, exhibited a higher number of haplotypes and greater diversity compared to BA.1* and BA.4*. This finding suggests that XBB* has the potential to become the predominant circulating variant in Vietnam in the future. The emergence and co-circulation of other Omicron CBVs, such as XBL, XAZ, and XBF, also mirror global epidemiological patterns observed elsewhere.
The findings from this study provide valuable insights into the ongoing epidemiological patterns of Omicron in Vietnam while highlighting its prevalence and diversity within different subvariants. These results align with existing literature on global Omicron dynamics [23,24], emphasizing the influence of globally circulating subvariants on local transmission patterns.
However, it is important to acknowledge some limitations of this study. First, the analysis was based solely on S gene sequences from GISAID database without considering other genomic regions or clinical data associated with these sequences. Incorporating additional genomic information and clinical metadata could provide a more comprehensive understanding of Omicron’s behavior in Vietnam. Furthermore, although haplotype analysis provides insights into genetic diversity within subvariants, it does not capture potential functional implications or phenotypic characteristics associated with specific haplotypes or mutations. Future research should aim to address these limitations by conducting more comprehension.
5. CONCLUSION
Our analysis reveals a high genetic diversity of Omicron variants in Vietnam with complex nucleotide changes in the S gene. Within the S gene, the S1 region exhibits a high accumulation of mutations, while the S2 region remains highly conserved across most Omicron subvariants. Our research also elucidates the genetic relationships among these Omicron subvariants, with BA.2* emerging as a central variant in the formation of the lineage comprising its offspring, including BA.4*, BA.5*, and XBB*.
6. FUNDING
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
7. AUTHORS’ CONTRIBUTIONS
All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; agreed to submit to the current journal; gave final approval of the version to be published; and agreed to be accountable for all aspects of the work. All the authors are eligible to be an author as per the International Committee of Medical Journal Editors (ICMJE) requirements/guidelines.
8. CONFLICTS OF INTEREST
The authors report no financial or any other conflicts of interest in this work.
9. ETHICAL APPROVALS
This study does not involve experiments on animals or human subjects.
10. DATA AVAILABILITY STATEMENT
All datasets were generated and analyzed in the present study.
11. USE OF ARTIFICIAL INTELLIGENCE (AI)-ASSISTED TECHNOLOGY
The authors declares that they have not used artificial intelligence (AI)-tools for writing and editing of the manuscript, and no images were manipulated using AI.
12. PUBLISHER’S NOTE
All claims expressed in this article are solely those of the authors and do not necessarily represent those of the publisher, the editors and the reviewers. This journal remains neutral with regard to jurisdictional claims in published institutional affiliation.
REFERENCES
1. V'kovski P, Kratzel A, Steiner S, Stalder H, Thiel V. Coronavirus biology and replication:implications for SARS-CoV-2. Nat Rev Microbiol 2021;19:155-70. [CrossRef]
2. Shahhosseini N, Babuadze GG, Wong G, Kobinger GP. Mutation signatures and in silico docking of novel SARS-CoV-2 variants of concern. Microorganisms 2021;9:926. [CrossRef]
3. Gowrisankar A, Priyanka TM, Banerjee S. Omicron:A mysterious variant of concern. Eur Phys J Plus 2022;137:100. [CrossRef]
4. Elliott P, Eales O, Bodinier B, Tang D, Wang H, Jonnerby J, et al. Dynamics of a national Omicron SARS-CoV-2 epidemic during January 2022 in England. Nat Commun 2022;13:4500. [CrossRef]
5. Callaway E, Ledford H. How bad is Omicron?What scientists know so far. Nature 2021;600:197-9. [CrossRef]
6. Duong BV, Larpruenrudee P, Fang T, Hossain SI, Saha SC, Gu Y, et al. Is the SARS CoV-2 omicron variant deadlier and more transmissible than Delta variant?Int J Environ Res Public Health 2022;19:4586. [CrossRef]
7. Allen H, Tessier E, Turner C, Anderson C, Blomquist P, Simons D, et al. Comparative transmission of SARS-CoV-2 Omicron (B.1.1.529) and Delta (B.1.617.2) variants and the impact of vaccination:National cohort study, England. Epidemiol Infect 2023;151:e58. [CrossRef]
8. Viana R, Moyo S, Amoako DG, Tegally H, Scheepers C, Althaus CL, et al. Rapid epidemic expansion of the SARS-CoV-2 Omicron variant in southern Africa. Nature 2022;603:679-86. [CrossRef]
9. Sun Y, Lin W, Dong W, Xu J. Origin and evolutionary analysis of the SARS-CoV-2 Omicron variant. Int J Biosaf Biosecur 2022;4: 33-7. [CrossRef]
10. Gu H, Krishnan P, Ng DY, Chang LD, Liu GY, Cheng SS, et al. Probable transmission of SARS-CoV-2 omicron variant in quarantine hotel, Hong Kong, China, november 2021. Emerg Infect Dis 2022;28:460-2. [CrossRef]
11. Poudel S, Ishak A, Perez-Fernandez J, Garcia E, León-Figueroa DA, RomaníL, et al. Highly mutated SARS-CoV-2 Omicron variant sparks significant concern among global experts - what is known so far?J Travel Med 2022;45:102234. [CrossRef]
12. Mohapatra RK, Kandi V, Sarangi AK, Verma S, Tuli HS, Chakraborty S, et al. The recently emerged BA.4 and BA.5 lineages of Omicron and their global health concerns amid the ongoing wave of COVID-19 pandemic - correspondence. Int J Surg 2022;103:106698. [CrossRef]
13. Tamura T, Ito J, Uriu K, Zahradnik J, Kida I, Anraku Y, et al. Virological characteristics of the SARS-CoV-2 XBB variant derived from recombination of two Omicron subvariants. Nat Commun 2023;14:2800. [CrossRef]
14. Parums DV. Editorial:The XBB.1.5 ('Kraken') subvariant of omicron SARS-CoV-2 and its rapid global spread. Med Sci Monit 2023;29:e939580. [CrossRef]
15. Thai PQ, Rabaa MA, Luong DH, Tan DQ, Quang TD, Quach HL, et al. The first 100 days of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) control in Vietnam. Clin Infect Dis 2021;72:e334-42. [CrossRef]
16. Wu F, Zhao S, Yu B, Chen YM, Wang W, Song ZG, et al.Anew coronavirus associated with human respiratory disease in China. Nature 2020;579:265-9. [CrossRef]
17. Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7:Improvements in performance and usability. Mol Biol Evol 2013;30:772-80. [CrossRef]
18. Tamura K, Stecher G, Kumar S. MEGA11:Molecular evolutionary genetics analysis version 11. Mol Biol Evol 2021;38:3022-7. [CrossRef]
19. Rozas J, Ferrer-Mata A, Sánchez-DelBarrio JC, Guirao-Rico S, Librado P, Ramos-Onsins SE, et al. DnaSP 6:DNA sequence polymorphism analysis of large data sets. Mol Biol Evol 2017;34: 3299-302. [CrossRef]
20. Minh BQ, Schmidt HA, Chernomor O, Schrempf D, Woodhams MD, von Haeseler A, et al. IQ-TREE 2:New models and efficient methods for phylogenetic inference in the genomic era. Mol Biol Evol 2020;37:1530-4. [CrossRef]
21. Leigh JW, Bryant D. Popart:Full-feature software for haplotype network construction. Methods Ecol Evol 2015;6:1110-6. [CrossRef]
22. Excoffier L, Lischer HE. Arlequin suite ver 3.5:A new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Resour 2010;10:564-7. [CrossRef]
23. Mohapatra RK, Kandi V, Verma S, Dhama K. Challenges of the omicron (B.1.1.529) variant and its lineages:A global perspective. Chembiochem 2022;23:e202200059. [CrossRef]
24. Islam MR, Shahriar M, Bhuiyan MA. The latest omicron BA.4 and BA.5 lineages are frowning toward COVID-19 preventive measures:A threat to global public health. Health Sci Rep 2022; 5:e884. [CrossRef]
25. Chavda VP, Balar P, Vaghela D, Solanki HK, Vaishnav A, Hala V, et al. Omicron variant of SARS-CoV-2:An Indian perspective of vaccination and management. Vaccines (Basel) 2023; 11:160. [CrossRef]
26. Ou J, Lan W, Wu X, Zhao T, Duan B, Yang P, et al. Tracking SARS-CoV-2 Omicron diverse spike gene mutations identifies multiple inter-variant recombination events. Signal Transduct Target Ther 2022;7:138. [CrossRef]
27. Zhao Z, Zhou J, Tian M, Huang M, Liu S, Xie Y, et al. Omicron SARS-CoV-2 mutations stabilize spike up-RBD conformation and lead to a non-RBM-binding monoclonal antibody escape. Nat Commun 2022;13:4958. [CrossRef]
28. Christopher R, Thomas PP, Luis Mariano P, Diego M, Maria Soledad A, Angie SH, et al. Mutational spectra distinguish SARS-CoV-2 replication niches. bioRxiv 2022;2022.09.27.509649. [CrossRef]
29. Quan TK, Phuoc H, Duc LM. Variations in spike gene of SARS-CoV-2 isolated in Vietnam. Int J Biosci 2022;21:83-92.
30. Quan TK, Phuoc H, Yen LT, Huyen NT. Genetic diversity of SARS-CoV-2 Omicron variants'spike gene in Vietnam. Int J Biosci 2022;21:166-76.
31. Qu P, Faraone JN, Evans JP, Zheng YM, Carlin C, Anghelina M, et al. Enhanced evasion of neutralizing antibody response by Omicron XBB.1.5, CH.1.1, and CA.3.1 variants. Cell Rep 2023;42: 112443. [CrossRef]
32. Hadfield J, Megill C, Bell SM, Huddleston J, Potter B, Callender C, et al. Nextstrain:Real-time tracking of pathogen evolution. Bioinform 2018;34:4121-3. [CrossRef]
33. Tegally H, Moir M, Everatt J, Giovanetti M, Scheepers C, Wilkinson E, et al. Emergence of SARS-CoV-2 omicron lineages BA.4 and BA.5 in South Africa. Nat Med 2022;28:1785-90. [CrossRef]
34. Sang P, Chen YQ, Liu MT, Wang YT, Yue T, Li Y, et al. Electrostatic interactions are the primary determinant of the binding affinity of SARS-CoV-2 Spike RBD to ACE2:A computational case study of omicron variants. Int J Mol Sci 2022;23:14796. [CrossRef]