
1. INTRODUCTION
Microbes are believed to contribute to the development of neurodegenerative diseases, including Alzheimer’s disease, Parkinson’s disease, and multiple sclerosis. The precise mechanisms underlying this association are still being investigated, but it is suggested that certain microbes may elicit an immune response in the brain, resulting in chronic inflammation and damage to neurons [1-3]. Several techniques have been used to identify microbial diseases in different kinds of diseases [Figure 1]. For instance, the bacterium Porphyromonas gingivalis, which is commonly associated with gum disease, can travel through the bloodstream and reach the brain, where it can promote the formation of amyloid plaques, a hallmark of Alzheimer’s disease, and damage neurons. Besides, some viruses and bacteria are also linked to the development of Parkinson’s disease. For instance, the herpes simplex virus has been detected in the brains of Parkinson’s patients, and certain pesticides have been shown to raise the risk of developing the disease in individuals who are infected [4,5]. While the relationship between microbes and neurodegenerative disease is complicated and not yet fully understood, ongoing research is providing novel insights into the potential role of microbes in the development and progression of these debilitating conditions. Alzheimer’s disease is a neurological disorder characterized by progressive cognitive decline, memory loss, and changes in behavior. With around 55 million individuals affected globally in 2020, Alzheimer’s disease is the primary cause of dementia in older adults. In 2021, the estimated total cost of care for individuals with Alzheimer’s disease and other dementias in the United States will be approximately $355 billion [6,7]. Over 6 million people across the United States are currently affected by Alzheimer’s disease, spanning across all age groups. According to projections for the year 2023, an estimated 6.7 million Americans aged 65 or older will be diagnosed with Alzheimer’s disease. A significant percentage of these individuals, around 73%, will be over the age of 75. It is estimated that by the year 2050, the global population of a particular demographic will increase to almost 13 million individuals [8].
 | Figure 1: Timeline of significant milestones in single-cell sequencing of microbial communities, along with the technique used up to 2023. [Click here to view] |
Research studies have increasingly suggested that microbial infections could be linked to the development and progression of Alzheimer’s disease. Studies have found that microbial agents such as bacteria, viruses, and fungi can be present in the brains of Alzheimer’s patients, contributing to the disease’s development [9-11]. Alzheimer’s disease pathogenesis is multifaceted, including genetic, environmental, and lifestyle factors. The disease is identified by an accumulation of amyloid beta (Aβ) and tau proteins in the brain, forming abnormal clumps and tangles. These clumps and tangles disrupt normal brain function and eventually lead to neuronal death [12-15]. Historically, Alois Alzheimer first described Alzheimer’s disease in the early 1900s [12,16]. At that time, a slow-acting virus was believed to be the cause, but this theory was later disproven. Present-day studies focus on the role of chronic infections in Alzheimer’s disease development [16]. Several microbial agents are believed to be involved in Alzheimer’s disease pathogenesis, including Porphyromonas gingivalis [4,17], Treponema denticola [18], Fusobacterium nucleatum [10], herpes simplex virus type 1 (HSV-1) [5], and Chlamydia pneumonia [19]. These agents enter in the brain through different mechanisms, including the blood-brain barrier and olfactory system, and cause inflammation and oxidative stress, leading to neuronal damage and the formation of amyloid plaques and tau tangles [5,20-22]. Recent studies have further strengthened the link between microbial infections and Alzheimer’s disease. For instance, a study in 2020 found that a fungal infection called Candida albicans could trigger the development of Alzheimer’s disease in mice. Another study in 2021 and 2022 found a connection between periodontal disease and an increased risk of developing Alzheimer’s disease [23,24].
Single-cell sequencing is a powerful tool for understanding the complex molecular mechanisms involved in Alzheimer’s disease and microbial infections. It allows for a comprehensive analysis of individual cells, providing a high-resolution view of the genetic and epigenetic changes that occur during infection and disease progression [25]. Single-cell sequencing can provide insights into the cellular interactions and pathways that contribute to disease pathogenesis. For instance, a study used single-cell sequencing to identify different immune-cell populations in the brains of Alzheimer’s patients, shedding new light on the immune system’s role in disease development [25]. Moreover, single-cell sequencing can help identify microbial agents present in the brains of Alzheimer’s patients. A study used single-cell sequencing to identify bacterial species in postmortem brain tissue from Alzheimer’s patients, which may help understand the role of microbes in disease pathogenesis. It also helps identify the molecular mechanisms underlying resistance to microbial infections and the effects of antimicrobial therapies. A study using single-cell sequencing to analyze immune cells from patients with tuberculosis revealed genetic changes associated with resistance to antimicrobial drugs. Overall, single-cell sequencing shows great promise for advancing our understanding of the complex molecular interactions involved in Alzheimer’s disease and microbial infections. It may help identify new therapeutic targets and develop personalized treatment strategies for this devastating disease [26,27]. Detailed experimental and computational approach steps are shown in Figure 2 and Table 1.
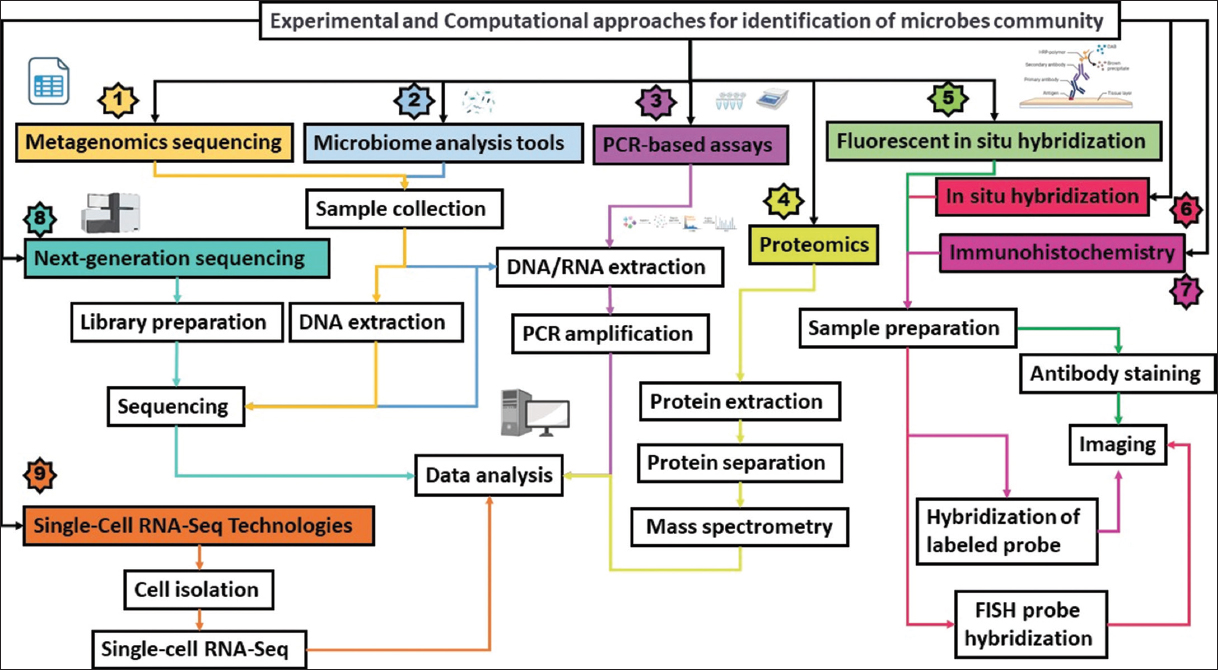 | Figure 2: Experimental and computational approaches for studying microbial pathogenesis in Alzheimer’s disease. The rectangular box is connected with the same color arrow that connects the final steps of techniques. [Click here to view] |
Table 1: Bioinformatics tools commonly used for studying microbial pathogenesis in Alzheimer’s disease.
| Tools | Use | Tool Links | References |
|---|---|---|---|
| QIIME (Quantitative Insights Into Microbial Ecology | analyze metagenomics data (Example: quality control, taxonomic classification, and diversity analysis) | http://qiime.org/ | [140] |
| MG-RAST | analyze metagenomics data (Example: quality control, taxonomic classification, functional annotation, and comparative analysis) | https://www.mg-rast.org/ | [141] |
| MetaPhlAn | profiling the microbial composition of metagenomic samples | https://huttenhower.sph.harvard.edu/metaphlan | [72] |
| HUMAnN2 | used for functional analysis of metagenomic data | https://huttenhower.sph.harvard.edu/humann/ | [142] |
| PathSeq | used for detecting microbial pathogens in metagenomic samples | https://software.broadinstitute.org/pathseq/ | [143] |
| MetaGOmics | analyze metagenomics data (quality control, taxonomic profiling, gene prediction, and functional annotation) | https://meta.yeastrc.org/metagomics/home.do | [144] |
| MEGAN | analyze metagenomics data (clustering, alignment, and visualization of taxonomic data) | http://ab.inf.uni-tuebingen.de/software/megan6/ | [145] |
| PICRUSt | functional analysis of microbial communities based on their 16S rRNA gene profiles | https://picrust.github.io/picrust/ | [146] |
| MetaBAT | binning metagenomic contigs into genomes | https://bitbucket.org/berkeleylab/metabat | [147] |
| Kraken | used for taxonomic classification of metagenomic reads | https://ccb.jhu.edu/software/kraken/ | [73] |
1.1. Correlation between Microbial Infections and Amyloid Beta (Aβ)
Recent investigations have uncovered a previously overlooked facet of amyloid beta (Aβ), a protein traditionally associated with Alzheimer’s disease pathology. Beyond its role in Alzheimer’s, Aβ has emerged as a substance with antimicrobial properties, suggesting a potential involvement in the brain’s innate immune system [28,29]. Studies indicate that Aβ exhibits noteworthy antibacterial and antifungal activities, implying a role in protecting the brain against diverse pathogens. The recognition of Aβ as an active participant in the innate immune response underscores its potential significance during infections, with increased Aβ production observed in response to microbial threats. This revelation prompts intriguing inquiries into the evolutionary context of Aβ, proposing that its antimicrobial properties might have evolved as a defense mechanism against infections. Nevertheless, the delicate equilibrium between Aβ production and clearance is pivotal. Dysregulation and excessive accumulation of Aβ are associated with neurodegenerative disorders, particularly Alzheimer’s disease. This evolving understanding of Aβ as an antimicrobial peptide not only sheds light on its potential protective functions but also opens new avenues for exploring the intricate dynamics between the immune system and neurodegenerative diseases. These insights may pave the way for novel therapeutic strategies in the realm of neurodegenerative research [28,29].
Predominantly focusing on humans, various studies have implicated specific microbes in the brains of the elderly, with notable mentions including herpes simplex virus type 1 (HSV1), Chlamydia pneumoniae, and various spirochaete types as potential contributors to the etiology of Alzheimer’s disease (AD) [5,19,30]. In addition, instances of fungal infection in AD brains have been documented [22], alongside reports of abnormal microbiota in the blood of AD patients. The initial observations of HSV1 in AD brains date back almost three decades. The growing body of research in this area, with approximately 100 studies solely focusing on HSV1, underscores the need for a reassessment of the infection and AD relationship. Recent investigations suggest a significant association between the gene encoding cholesterol 25-hydroxylase (CH25H) and the development of Alzheimer’s disease (AD). Virus infection selectively triggers the upregulation of CH25H, leading to the generation of its enzymatic product, 25-hydroxycholesterol (25OHC), which activates innate antiviral immunity [31,32]. Polymorphisms within human CH25H are implicated in both AD susceptibility and amyloid-beta (Aβ) deposition, suggesting that Aβ induction may be a key target of 25OHC. This provides a potential mechanistic link between infection and Aβ production [29,33]. Moreover, Aβ itself is recognized as an antimicrobial peptide with potent activity against various bacteria and yeast, underscoring its role in the immune response. This introduces another layer to the possible connection between infections and AD, as Aβ also demonstrates antiviral activity [29,34-36]. In addition, the heightened expression of another antimicrobial peptide, β-defensin 1, in the AD brain further supports the correlation between the immune system’s response to infections and the pathological processes observed in Alzheimer’s disease [37].
1.2. Primary Advantages of Single-cell Sequencing Over Bulk Sequencing
Single-cell sequencing is a revolutionary technology that enables researchers to study individual cells in isolation, as opposed to traditional bulk sequencing methods that measure the genetic information of an entire population of cells simultaneously. The technology provides new avenues for research by allowing researchers to study cellular heterogeneity, identify rare cell types, and characterize the genetic and epigenetic landscape of individual cells. Single-cell sequencing involves isolating individual cells and amplifying their genetic material before sequencing it. Different types of single-cell sequencing technologies, such as RNA sequencing (scRNA-seq), DNA sequencing (scDNA-seq), and chromatin accessibility profiling (scATAC-seq), are available [38-40].
The primary advantages of single-cell sequencing over bulk sequencing are as follows:
Increased resolution: Single-cell sequencing provides researchers with the ability to study individual cells, enabling them to identify rare cell types and reveal cellular heterogeneity that would have been missed with bulk sequencing [41].
Accurate representation of biology: Traditional bulk sequencing techniques tend to obscure the true biology of a sample by averaging the genetic information of all cells within the sample. In contrast, single-cell sequencing provides a more accurate representation of the biology of a sample by allowing researchers to study the genetic information of each cell [42,43].
Identification of rare cells: Single-cell sequencing is an effective tool for identifying rare cell types present in low abundance in a sample. Such rare cells can be critical for understanding disease pathology or developing new therapies [44].
Understanding of cellular development: Single-cell sequencing can help researchers gain insights into how cells develop and differentiate into various cell types [45,46].
One significant example of the power of single-cell sequencing is its use in cancer research. By studying individual cancer cells, researchers can identify genetic mutations and epigenetic changes that are specific to individual cells. This enables them to understand the evolution of cancer cells over time and identify novel therapeutic targets [47]. Recently, researchers used single-cell sequencing to study the genetic heterogeneity of bladder cancer. They identified multiple subpopulations of cancer cells with distinct genetic and epigenetic features and mapped the developmental trajectory of the cancer cells. The study provides new insights into the development of bladder cancer and could lead to the development of more effective therapies [48-50].
1.3. Single-cell Sequencing Techniques to Identify Potential Pathogens in Alzheimer’s Disease Patients
Alzheimer’s disease (AD) is a neurological disorder that affects memory cognitive function and ultimately leads to neuronal death [12,51,52]. Recent studies have shown that there may be a link between certain pathogens and the development of Alzheimer’s disease (AD) [11,17,18,22,53-55] [Figure 3, Table 2]. Various research studies have utilized single-cell sequencing techniques to identify potential pathogens in AD patients. One study published in Frontiers in Molecular Neuroscience analyzed brain samples from AD patients and non-demented controls using single-cell RNA sequencing (scRNA-seq) [56-58]. The researchers observed an upregulation of genes related to the herpes simplex virus type 1 (HSV-1) in AD patients compared to controls [5]. They also found a correlation between the level of HSV-1 expression and the severity of AD symptoms. This study suggests that HSV-1 may play a role in the development of AD [5]. Li et al., identified several viral RNA sequences in brain tissue samples from AD patients, including herpesviruses and human endogenous retroviruses. They also found that the expression of these viral sequences was correlated with the expression of genes associated with neuroinflammation and AD pathology [27]. scRNA-seq was used to explore the potential role of the oral microbiome in AD. The researchers found oral bacteria present in brain tissue samples from AD patients, suggesting that the oral microbiome may be involved in the development of AD. Overall, these studies provide evidence to suggest that infectious agents, such as viruses and oral bacteria, may contribute to the development of AD. Further research is required to fully comprehend the relationship between pathogens and AD and to develop new treatments and prevention strategies [4,17,27].
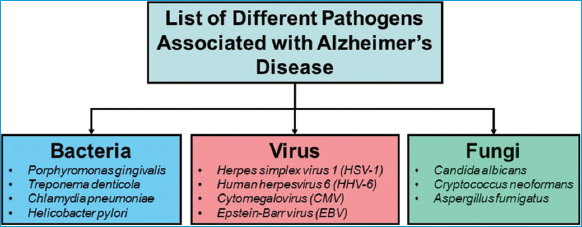 | Figure 3: Potential bacteria, viruses, and fungal pathogens associated with AD development and progression. [Click here to view] |
Table 2: Potential Pathogens Linked to Alzheimer’s Disease Development and Progression.
| Microbes | Effects in AD | References |
|---|---|---|
| Herpes simplex virus (HSV) | increased risk o f Alzheimer’s disease | [5] |
| Porphyromonas gingivalis | cause neurodegeneration in mice | [4,17] |
| Chlamydia pneumonia | increased risk of Alzheimer’s disease | [19] |
| Spirochetes | cause neurodegeneration in mice | [20] |
| Streptococcus | neurodegeneration in mice | [148] |
| Bacteroides fragilis | neurotoxic proteins | [149] |
| Enterococcus faecalis | formation of amyloid beta plaques | [29] |
| Escherichia coli | contribute to neuroinflammation | [64] |
| Human herpesvirus 6A and 7 | increased risk of Alzheimer’s disease | [150] |
| Fungi (Saccharomyces cerevisiae, Malassezia globosa, Malassezia restricta, Penicillium, Phoma) | neurodegeneration in mice | [22] |
| Candida tropicalis, Candida krusei | [22] | |
| Aspergillus fumigatus, Cryptococcus neoformans, and Malassezia spp | AD pathogenesis | [151] |
Pettas et al. utilized single-cell RNA sequencing to examine the transcriptional profiles of brain cells in Alzheimer’s patients. The study revealed a network of genes involved in antiviral immunity that was disrupted in Alzheimer’s patients compared to healthy controls. The study also observed evidence of viral infection in the brains of Alzheimer’s patients, including the presence of viral RNA and protein. These findings suggest that viral infection might contribute to the neuroinflammation and neuronal damage that occur in Alzheimer’s disease [58,59]. Another study utilized single-cell sequencing to investigate the role of the gut microbiome in Alzheimer’s disease. The study observed that the gut microbiome of Alzheimer’s patients was distinct from that of healthy controls, with an increase in pro-inflammatory bacteria and a decrease in anti-inflammatory bacteria. The study also found evidence of gut bacterial translocation to the brain, suggesting that the gut-brain axis might be involved in Alzheimer’s disease pathogenesis [2,60,61]. The authors have made a significant breakthrough in delivering genetic material into microglia cells using newly discovered adeno-associated virus (AAV) variants. Microglia cells are crucial immune cells that shape neural circuits in the CNS and play a vital role in immune responses. The AAV-cMG and AAV-MG variants can efficiently deliver genetic payloads, including fluorescent labeling, calcium and neurotransmitter imaging, and genome editing in microglia in vivo. The discovery of these AAV variants is expected to aid in the study of microglia biology and the underlying mechanisms of diseases associated with microglial dysfunction without inducing immune activation in microglia cells [62].
Recent studies have suggested a potential link between the gut microbiome and the development and progression of AD. Single-cell sequencing (SCS) is a technique that has the potential to identify specific microbial species involved in AD. SCS allows for the high-resolution profiling of the microbiome at the single-cell level, which can reveal rare microbial populations and identify individual microbes within complex communities. By providing detailed information about the genetic makeup and functional capacity of individual microbial cells, SCS can help researchers investigate their roles in disease pathogenesis [2,62-64]. One study used SCS to analyze the gut microbiome of AD patients and healthy controls and identified differences in the abundance of certain microbial species between the two groups. The researchers found an increase in pro-inflammatory bacteria, such as Escherichia coli and Shigella flexneri, in AD patients compared to controls, while anti-inflammatory bacteria like Faecalibacterium prausnitzii were reduced in AD patients [65,66]. Another study used SCS to investigate the oral microbiome of AD patients and found an increase in the abundance of oral bacteria like Porphyromonas gingivalis in AD patients compared to healthy controls. Porphyromonas gingivalis is known to produce amyloid beta, a key pathological feature of AD, and its presence in the brain has been linked to an increased risk of developing AD [4,10,55,67]. These findings suggest that SCS can be a valuable tool for identifying specific microbial species involved in AD. However, more research is needed to confirm these results and to elucidate the mechanisms by which these microbial species contribute to AD pathogenesis.
Single-cell sequencing (SCS) has emerged as a powerful technique for identifying specific microbial species involved in Alzheimer’s disease (AD). Several recent experimental studies have utilized SCS to investigate the dysbiosis of the gut and oral microbiomes in AD patients [65]. There is an increased abundance of several pro-inflammatory bacterial species, such as Escherichia coli, Shigella flexneri, and Ruminococcus torques, in AD patients compared to controls [60]. Furthermore, AD patients showed a reduced abundance of anti-inflammatory bacterial species, including Faecalibacterium prausnitzii and Eubacterium rectale [64,66]. These findings suggest that gut dysbiosis and inflammation may play a role in the development and progression of AD [65]. Soreq et al., investigated the oral microbiome of AD patients and healthy controls using SCS and they found that AD patients had a significantly higher abundance of several oral bacterial species, including Actinomyces naeslundii, Tannerella forsythia, and Porphyromonas gingivalis, compared to controls [23,54]. Porphyromonas gingivalis is known to produce amyloid beta, a key pathological feature of AD, and its presence in the brain has been linked to an increased risk of developing the disease. These findings suggest that the oral microbiome may also contribute to AD pathogenesis [17,60]. Taken together, these studies demonstrate the potential of SCS to identify specific microbial species involved in AD and provide important insights into the dysbiosis of the gut and oral microbiomes in AD patients [65].
1.4. Limitations and Challenges in Using Single-cell Sequencing to Study Microbial Infections in Alzheimer’s Disease
Single-cell sequencing (SCS) is an effective tool for exploring microbial infections in Alzheimer’s disease (AD). However, several challenges can arise when using this technique. These challenges include limited sensitivity, difficulty in distinguishing microbial species, lack of reference databases, contamination, cost, and sample size and diversity [Figure 4]. These challenges can impact the quality of SCS data and, therefore, the accuracy of microbial identification in AD. To address these challenges, researchers can use various strategies. For example, they can use FISH or immunofluorescence to identify microbial cells before SCS, which can improve the sensitivity of the technique. In addition, bioinformatics tools such as metagenomics analysis and marker genes can assist in the identification of microbial species. Researchers can also use negative controls to monitor contamination during sample preparation and sequencing, and they can consider pooling cells from multiple samples to reduce costs. Overall, while SCS has limitations, careful experimental design, analytical approaches, and the use of emerging technologies can help overcome these challenges and provide valuable insights into the microbial communities in AD.
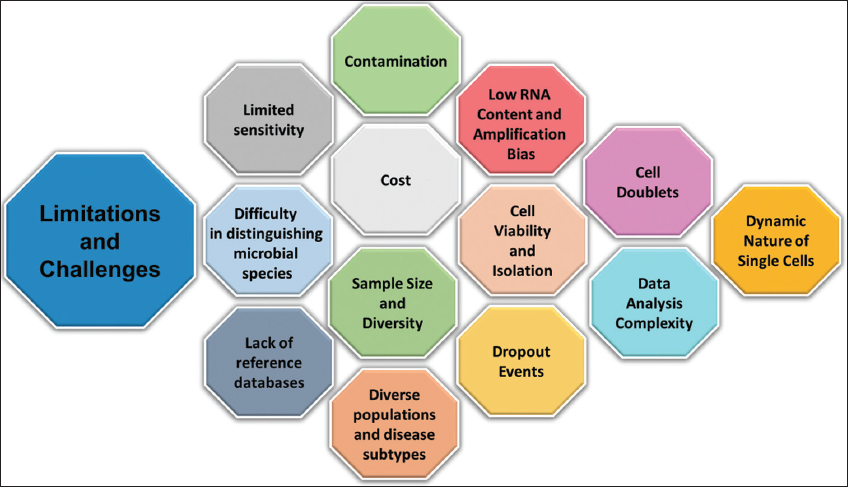 | Figure 4: Limitations and challenges associated with Single-cell sequencing. [Click here to view] |
1.5. Limited Sensitivity
Single-cell sequencing (SCS) requires high-quality RNA from individual cells, which can be challenging to obtain from infected tissues. In addition, some microbes may be present at low frequencies, making their detection through SCS difficult. Researchers can address this limitation using techniques such as fluorescence in situ hybridization (FISH) or immunofluorescence to visualize microbial cells before isolating them for SCS. Moreover, emerging technologies such as droplet-based SCS or single-nucleus RNA sequencing (snRNA-seq) can improve the sensitivity of SCS by enabling the analysis of larger numbers of cells. For instance, in a study by [23,68], the researchers used FISH to increase the microbial cell frequency in the oral cavity of patients with periodontitis. Brase et al. conducted a single-nucleus RNA sequencing (snRNA-seq) study to investigate gene expression changes in Alzheimer’s disease [69]. The researchers detected differences in gene expression between Alzheimer’s patients and healthy controls despite low-quality RNA samples.
1.6. Difficulty in Distinguishing Microbial Species
SCS can generate vast amounts of data, and analyzing this data to identify specific microbial species can be challenging. Furthermore, some microbes may have similar genomic sequences, making it difficult to differentiate them from one another. Researchers can tackle this issue by using bioinformatics tools such as metagenomics analysis, which compares the SCS data to reference databases to identify microbial species. In addition, researchers can use complementary techniques such as shotgun metagenomics or 16S rRNA sequencing to obtain more information on the microbial community [70]. For example, Lloyd-Price et al. used metagenomics analysis to investigate the gut microbiota of patients with inflammatory bowel disease [71].
1.7. Lack of Reference Databases
Some microbial species are not yet well-characterized, and there may be limited information available in reference databases, which can make it difficult to assign taxonomic identities to microbial sequences identified using SCS. To address this limitation, researchers can use tools such as Kraken, which uses k-mer analysis to identify microbial species, or MetaPhlAn, which uses marker genes to identify microorganisms [72,73]. In addition, researchers can create custom reference databases for their specific study systems. For example, in a study by Rieder et al. (2023), the researchers developed a custom reference database to investigate the skin microbiome of patients with atopic dermatitis [74].
1.8. Contamination
Contamination of samples with environmental or laboratory microbes can result in false-positive results. To minimize the risk of contamination, researchers must carefully design experiments and analyze the data. Negative controls, such as sterile water, can be used to monitor for contamination during sample preparation and sequencing. Computational tools such as Decontam can identify and remove potential contaminants from SCS data. For example, Gryaznova et al. used negative controls and Decontam to investigate the microbial community in the vaginal tract of pregnant women [75]. Bao et al. used negative controls and computational tools to investigate the oral microbiota of patients with periodontitis. The study identified several microbial species associated with periodontitis, such as Porphyromonas gingivalis, and distinguished true bacterial cells from potential contaminants [18,54]. Incomplete digestion of genomic DNA (gDNA) during total RNA extraction, the presence of doublets or multiplets (multiple cells grouped together), ambient contamination from nucleic acids released by dying cells, and the inclusion of cell-free mRNA (ambient RNA) can all introduce confounding factors. In addition, the infiltration of normal cells surrounding tumors can contaminate expression data obtained from tumor samples. Vigilance against these contamination sources is crucial for accurate scRNA-seq analysis. Droplet-based single-cell RNA-seq (scRNA-seq) data often suffer from ambient contamination due to nucleic acid material from deceased cells. This material mixes into the buffer and is co-encapsulated with cells, reducing the signal-to-noise ratio [76].
Ambient RNA contamination diminishes sequencing read depth and introduces a misleading signal, obscuring biological insights. Tissue dissociation in single-cell suspensions is a known source of contamination. Various strategies exist, such as van der Wijst et al.’s collagenase protocol for gut mucosal biopsies. While these protocols enhance cell viability, they neglect ongoing stresses post-dissociation. Single cells in suspension are prone to death, prompting the development of single-nucleus RNA-seq (snRNA-seq) for death-prone tissues. However, isolated nuclei carry cytoplasmic RNA and ribosomes, and lysis buffers may cause RNA leakage. Droplet-based approaches often overlook the impact of fluidic technologies [77-80]. DecontX introduces an innovative Bayesian approach for precise estimation and elimination of contamination in individual cells. It successfully forecasts contamination levels in mixed mouse-human datasets and rectifies abnormal expression of marker genes in PBMC datasets [81]. Introducing SoupX, a cutting-edge solution designed to eliminate ambient RNA interference in droplet-based single-cell RNA sequencing endeavors. With versatile applications, its implementation enhances the biological relevance of both current and forthcoming datasets [82]. In the realm of single-cell RNA sequencing (scRNA-seq), several robust tools play pivotal roles in addressing contamination and enhancing data quality. SoupX stands out as a versatile solution adept at eliminating ambient RNA interference, thereby significantly improving the biological relevance of scRNA-seq datasets. In addition, Scrublet proves invaluable in identifying and filtering out both ambient RNA and doublets, contributing to heightened data accuracy [83]. The tool DoubletFinder specializes in identifying and flagging potential doublets within scRNA-seq data, ensuring the purity of single-cell populations. For tackling broader issues such as batch effects and unwanted variation, RUV-III emerges as a powerful tool, effectively reducing non-biological sources of variability and thus enhancing data quality [84,85]. Moreover, DecontX plays a crucial role in detecting and removing contaminating cell types, contributing to the specificity of scRNA-seq data by eliminating unwanted cellular elements. Incorporating these tools into scRNA-seq workflows equips researchers with comprehensive solutions for effective preprocessing, ensuring the reliability and accuracy of subsequent analyses [81]. The presence of cell doublets (two cells sequenced as one) and contamination with ambient RNA can confound the interpretation of scRNA-seq data. In the realm of single-cell RNA sequencing (scRNA-seq), the emergence of doublets arises when two cells are inadvertently combined in a single reaction volume. These doublets, although visually resembling real cells, introduce a significant challenge in scRNA-seq data analysis. While various computational techniques aim to identify doublets in scRNA-seq data, the absence of thorough benchmarking across these methods complicates the selection process for researchers seeking suitable approaches for their analyses [86].
1.9. Cost
The significant expense associated with single-cell RNA sequencing (scRNA-seq) poses a notable challenge in genomics. The intricacies of scRNA-seq experiments, including library preparation, sequencing, and data analysis, contribute to high costs. Specialized equipment, reagents, and skilled personnel further inflate expenses, limiting the scope of studies. Addressing cost challenges is vital for broader access to scRNA-seq, promoting research in single-cell genomics, and advancing understanding of cellular heterogeneity and gene expression. Optimization of protocols, cost reduction in reagents, and the development of more economical platforms are essential for overcoming this obstacle and democratizing access to scRNA-seq benefits [87-90]. SCS is a relatively expensive technique compared to other sequencing methods, which may make it impractical for some research groups or projects. However, the cost of SCS has decreased over time, and new technologies such as droplet-based SCS have reduced the cost per cell [68]. Researchers can also consider pooling cells from multiple samples to reduce the overall cost. For example, pooled cells from multiple soil samples and used droplet-based SCS to investigate the diversity of microbial species in the soil. Droplet-based single-cell RNA sequencing is used to investigate the microbial communities in the lungs of patients with cystic fibrosis. The study identified specific microbial species associated with disease severity, such as Pseudomonas aeruginosa, at a lower cost than traditional SCS methods [68,91].
1.10. Sample Size and Diversity
Single-cell RNA sequencing (scRNA-seq), a transformative technology for exploring cellular heterogeneity, grapples with two major limitations: restricted sample size and inadequate diversity. Limited starting material poses challenges in detecting subtle gene expression differences and understanding biological variability, potentially compromising statistical power and result in reproducibility. In addition, scRNA-seq may inadvertently underrepresent certain cell types due to biases in isolation and profiling techniques, hindering a comprehensive understanding of the cellular landscape. To mitigate these issues, researchers can consider pooling samples, integrating multiple datasets, improving cell isolation techniques, and standardizing protocols. These efforts are crucial for enhancing statistical robustness, ensuring broader applicability of findings, and fully realizing the potential of scRNA-seq in elucidating the intricacies of biological systems. Soreq et al. conducted single-cell RNA-Seq analysis on frontal cortex samples obtained from two individuals with Alzheimer’s disease (AD) and two control subjects. Despite the limited dataset, the study reveals heightened levels of cell-specific markers associated with glial cells in Alzheimer’s samples compared to controls. The study’s design, limited by a single time point and a small sample size, presents challenges in determining whether the observed alterations signify upstream events in the disease’s pathogenesis or downstream consequences of neurodegeneration. In addition, the analysis identified an increased frequency of microglia, astrocytes, and oligodendrocytes, consistent with previous findings in AD reported in genome-wide transcriptome studies. However, the precise temporal positioning of these changes in relation to disease progression—whether they are upstream or downstream—remains undetermined. This uncertainty represents a potential limitation in interpreting the study’s findings [25].
1.11. Low RNA Content and Amplification Bias
Single cells contain minimal amounts of RNA, making it challenging to obtain sufficient material for sequencing. In addition, the amplification step introduces biases, leading to a distorted representation of gene expression profiles. Amplification bias may result in the over-representation of highly expressed genes and the loss of information from lowly expressed genes [92,93]. In single-cell RNA sequencing (scRNA-seq), low RNA count refers to the challenge of obtaining reliable gene expression profiles due to limited RNA content in individual cells. This limitation can arise from various factors and impact the accuracy of downstream analyses. To address and identify low RNA counts in scRNA-seq data, several tools and procedures are employed. Tools such as Scater and Seurat [44,94] offer functionalities for quality control, including the identification of cells with low RNA counts. The identification process involves analyzing gene expression distributions, examining library size or total counts per cell, and identifying genes with consistently low expression. Statistical metrics, like median absolute deviation (MAD) or Z-scores, are also employed to flag cells with significantly different expression profiles. Incorporating these tools and procedures into scRNA-seq analyses enables researchers to pinpoint cells with low RNA counts, facilitating improved quality control and enhancing the reliability of downstream biological interpretations. In scRNA-seq analysis, the initial step involves eliminating unlikely, intact individual cell barcodes. For high-throughput methods, a crucial stage is filtering out non-representative cell barcodes. One approach is to set a dataset-specific threshold based on the minimum number of UMIs required for considering a barcode as a cell. Alternatively, tools like EmptyDrops estimate background RNA levels in empty wells or droplets, identifying cell barcodes deviating significantly from the background and indicating cell presence. This strategy detects cell types with lower RNA content compared to others in the sample [95,96]. When analyzing samples with high ambient RNA percentages, DropletQC may overlook some damaged cells and empty droplets. However, these can be detected by their low RNA content and flagged using a minimum UMI threshold. It is advised to use DropletQC alongside tools like EmptyDrops to eliminate most cell barcodes before identifying any remaining damaged cells or cell-free droplets. The DropletQC package includes separate functions for calculating the nuclear fraction, identifying empty droplets, and detecting damaged cells [95,97].
Amplification bias in single-cell RNA sequencing (scRNA-seq) is a notable challenge as it distorts the accurate representation of gene expression, especially in scenarios with limited input material. This bias introduces errors during the identification of expressed genes, compromising data integrity. To counteract this issue, various tools and strategies come into play. Duplex sequencing (DuplexSeq) mitigates PCR-induced errors, enhancing precision. Unique molecular identifiers (UMIs) employ distinctive barcodes before amplification for accurate quantification, facilitated by tools such as UMI-tools. The identification of genome regions with reduced coverage, indicative of bias, is accomplished through allelic dropout analysis (ADS-seq). Moreover, incorporating external RNA control consortium (ERCC) spike-ins during library preparation enables the assessment and correction of amplification bias. The comprehensive identification procedure involves pre-processing steps, UMI handling, potential duplex sequencing, ERCC spike-in analysis, allelic dropout analysis, and the application of statistical methods for quantification and bias correction. Integrating these methodologies into the analytical pipeline ensures precise evaluation of gene expression in scRNA-seq, significantly enhancing the reliability of downstream analyses [98-102].
1.12. Cell Viability and Isolation
Isolating single cells while maintaining their viability is a delicate process. Cell stress or death during isolation can introduce artifacts in gene expression profiles. Effectively disassembling intricate systems like tissues or multicellular spheroids poses a considerable technical challenge. The use of mechanical or enzymatic approaches can result in cellular damage and may influence gene expression when executed too forcefully or for prolonged durations [103]. Hence, the identification of an appropriate dissociation methodology is of paramount importance. Regardless of the chosen method, three key criteria must be met for successful tissue dissociation: (1) achieving high cell recovery; (2) preserving cell integrity and functionality; and (3) employing a straightforward and replicable technique. In the context of techniques such as fluorescent-assisted cell sorting (FACS) and single-cell RNA sequencing (scRNA-seq), it is essential to convert microtissues into a single-cell suspension without compromising cellular integrity and minimizing alterations in gene expression. As a result, a predominant approach in research has involved employing enzymatic dissociation to investigate individual cell populations within the central nervous system (CNS) at a single-cell resolution. Nevertheless, recent findings in diverse tissues and cell types have shed light on potential artifacts arising from enzymatic digestion processes [104-106]. While two prior single-cell RNA sequencing (scRNA-seq) studies focused on the brain, the absence of biological replicates and/or limited representation of cells from less common types, such as microglia, has hindered the comprehensive, reproducible comparative analysis of the dissociation signature in the CNS [107,108].
1.13. Dropout Events
scRNA-seq can suffer from “dropout events,” where certain genes are not detected in individual cells, leading to incomplete expression profiles [109]. scRNA-seq investigations frequently yield substantial datasets, encompassing comprehensive gene expression measurements across thousands or more individual cells. This abundance of data poses significant computational challenges in analyzing and deciphering the information. Numerous factors contribute to the complexity of computational analysis for scRNA-seq data, including its high dimensionality, inherent measurement noise, detection limits, and the imbalanced size ratio between rare and abundant cell populations. Among the complexities, a notable feature influencing these challenges is the phenomenon termed “dropout,” where a gene exhibits a low or moderate expression level in one cell but remains undetected in another cell of the same type. These dropout occurrences stem from factors such as limited mRNA quantities in individual cells, inefficient mRNA capture, and the inherent stochasticity of mRNA expression. Consequently, the scRNA-seq data frequently manifest as highly sparse, with an overabundance of zero counts that renders it zero-inflated, capturing only a fraction of each cell’s transcriptome [110,111].
Due to the limited initial material available, single-cell RNA sequencing (scRNA-seq) encounters challenges associated with low capture efficiency and elevated dropouts, as highlighted by Haque et al. in 2017. In contrast to bulk RNA-seq, scRNA-seq yields data characterized by increased noise and variability. The presence of technical noise and biological variations, such as stochastic transcription, poses significant hurdles for the computational analysis of scRNA-seq data [89]. Single-cell RNA sequencing (scRNA-seq) data often exhibits missing values or dropouts, resulting from unsuccessful amplification of original RNAs. The occurrence of dropout events in scRNA-seq is influenced by the protocol used and correlates with the number of sequencing reads per cell [112]. These dropouts contribute to increased cell-to-cell variability, impacting the signal for each gene and complicating the detection of gene-gene relationships. Consequently, downstream analyses are significantly affected, as a considerable portion of genuinely expressed transcripts may go undetected in scRNA-seq. To address this issue, imputation emerges as a valuable strategy to replace missing data by introducing substituted values. It is worth noting that existing imputation methods for bulk RNA-seq data may not directly apply to scRNA-seq data [113].
1.14. Data Analysis Complexity
The intricacy of data analysis in single-cell RNA sequencing (scRNA-seq) stems from multiple factors. The process generates extensive datasets with individual cell gene expression profiles, resulting in high-dimensional data that demands advanced computational methods for interpretation. Technical challenges, such as RNA molecule capture efficiency and the presence of ambient RNA, contribute noise and variability to scRNA-seq data, requiring meticulous handling for accurate downstream analysis. Furthermore, the inherent heterogeneity in cell populations adds another layer of complexity. Advanced clustering and classification algorithms are necessary to identify and characterize distinct cell types or states within the dataset. In addition, understanding gene expression dynamics at the single-cell level involves employing specialized methods to model temporal patterns.
To address these challenges, researchers adopt a multi-step approach. Initial steps include preprocessing raw data, utilizing techniques like principal component analysis (PCA) and uniform manifold approximation and projection (UMAP) for dimensionality reduction, and employing clustering to delineate cell types or states [114,115]. Subsequent analysis involves tasks like differential expression analysis, trajectory reconstruction, and integrating multiple datasets. Visualization tools, such as ggplot2 and interactive platforms, play a crucial role in effectively presenting results. Effectively managing the complexity of data analysis in scRNA-seq requires staying abreast of evolving tools, leveraging parallel computing resources, collaborating with experts, and ensuring thorough documentation for reproducibility. These efforts contribute to continual enhancements in the methodologies employed for scRNA-seq analysis.
1.14.1. Dynamic nature of single cells
Single cells are dynamic, and their gene expression profiles can change rapidly. Capturing this dynamic nature poses a challenge in obtaining a snapshot that accurately represents the cell’s state. The concept of ‘cell state transition’ encompasses the dynamic changes cells undergo over time. This phenomenon is integral to embryonic development, where cells differentiate progressively. In addition, it plays a key role in maintaining tissue function during homeostasis and repair, replacing damaged cells. An understanding of these transitions is pivotal, as various disorders, spanning developmental issues to cancer, often result from abnormal shifts in cell states [116].
1.14.2. Batch effects
Variability introduced during different experimental batches can confound the interpretation of results. Batch effects in single-cell RNA sequencing (scRNA-seq) represent systematic variations in gene expression resulting from technical disparities between batches of samples processed in distinct experimental runs. These variations can obscure genuine biological signals, posing a challenge to distinguishing between true cellular differences and technical artifacts. Addressing batch effects is essential for the accurate interpretation of scRNA-seq data. Strategies to mitigate these effects include randomization and balancing in experimental design, normalization techniques such as global scaling and library size normalization, integration methods like Harmony and ComBat, as well as pseudo-bulk analysis. Utilizing quality control measures and filtering low-quality cells and features further aids in minimizing the impact of batch effects. Continuous advancements in computational methods contribute to enhancing the robustness of batch correction in scRNA-seq analyses [117-119]
1.15. Heterogeneity of Alzheimer’s Disease (AD) Across Diverse Populations and Disease Subtypes
The applicability of insights derived from single-cell RNA sequencing (scRNA-seq) in Alzheimer’s disease research depends on addressing the challenges associated with diverse populations and disease subtypes [120,121]. scRNA-seq has proven valuable in exploring the diversity of microglia within the human brain, addressing the challenge of heterogeneity. Recent examinations using single-cell RNA sequencing (scRNA-seq) on microglia in the human brain have revealed distinct variations within the microglial population. Sankowski et al. identified eight distinct clusters of microglia in the human brain, labeled C1–C3, C5–C9, each exhibiting unique gene expressions. They investigated the expression of these cluster-specific genes within myeloid cell populations using our scRNA-seq composite dataset [122-124]. ScRNA-seq offers a significant benefit by impartially pinpointing cellular subgroups within diverse cell populations. The intricate structure of tissues is intricately tied to their function, underscoring the vital importance of accurately discerning cell types and their frequencies [125]. Single-cell RNA-Seq offers the ability to uncover transcript heterogeneity at the single nucleotide level and identify gene expression diversity within individual cells. In contrast, analyzing cell populations provides only an averaged gene expression level. Recent research highlights that pooling results from 30 to 100 single cells can effectively reconstruct the averaged gene expression observed in the entire cell population, addressing stochastic gene expression heterogeneity. While our findings align with this concept, it is important to note that pooling just five cells from diverse treatments or conditions is insufficient for accurately representing the averaged expression of populations [126,127]. scRNA-seq information displays notable diversity, suggesting the presence of organized low-rank submatrices. In addition, a prior investigation indicated that variations in gene expression significantly influence dropout occurrences [111]. An expanding body of evidence reveals Alzheimer’s disease (AD) as a complex condition, diverging from traditional perspectives. Approximately one-third of clinically diagnosed AD patients lack Aβ accumulation and postmortem cases show cognitive normality. In sporadic late-onset AD, individuals may possess unique genetic variations such as CLU, TREM2, and APOE, heightening disease risk. The interplay between these genetic factors and disease progression remains unclear. Predicting AD progression is daunting, emphasizing significant heterogeneity in patient outcomes [128-130]. While single-cell omics have shown significant advancements, it may fall short in comprehending cellular diversity. Analyzing differential expression in scRNA-seq data to pinpoint cell-specific genes provides insights, but decoding cellular identity solely from gene regulation lists proves challenging. This difficulty arises because gene functions and the impact of disease-related variants hinge on their interactions within the cellular environment, emphasizing the importance of considering these relationships for a comprehensive understanding [131,132]. Alzheimer’s disease is recognized for its multifaceted nature, both clinically and biologically, prompting investigations into gene expression patterns at the cellular level using scRNA-seq. Ensuring the generalizability of these findings requires a nuanced understanding of how genetic, environmental, and lifestyle factors contribute to the disease across diverse populations. Likewise, the existence of various Alzheimer’s subtypes, such as early-onset and late-onset forms, as well as distinctions based on protein aggregates like amyloid-beta and tau, introduces complexities in interpreting scRNA-seq results across different subgroups. Technical intricacies in laboratory procedures and bioinformatics analyses, coupled with the imperative for validation in independent cohorts, shape the reliability and generalizability of scRNA-seq outcomes. Incorporating multi-omics data, encompassing genomics and proteomics, is a common strategy, demanding careful integration to ensure a cohesive interpretation across diverse data types. Continuous collaboration and advancements within the research community contribute to refining the uniqueness and applicability of scRNA-seq insights in understanding Alzheimer’s disease.
1.16. Potential Future Directions for Using Single-cell Sequencing to Investigate Microbial Infections
Single-cell sequencing is a cutting-edge technology that enables the analysis of individual cells within a complex and heterogeneous population, providing novel insights into the genetic and phenotypic diversity of microbial communities. In the context of Alzheimer’s disease, there are several potential future directions for using single-cell sequencing to investigate microbial infections.
Identification of microbial communities: Single-cell sequencing (SCS) can assist in identifying microbial species and strains present in Alzheimer’s disease (AD) brains. By studying the composition and dynamics of microbial communities, researchers can gain a better understanding of the role these communities play in AD pathogenesis.
Investigation of host-microbe interactions: SCS can be used to investigate the interactions between microbial communities and host cells in AD brains, identifying genes and pathways activated in response to microbial infection and their role in AD pathogenesis.
Identification of novel microbial targets: SCS can help identify specific microbial species or strains associated with AD pathology, enabling the development of therapies that target these microbes directly.
Evaluation of microbial therapies: SCS can evaluate the efficacy of microbial therapies in AD by monitoring the effects of interventions on microbial communities within the AD brain.
Single-cell sequencing can be utilized to identify rare or uncultivable microorganisms that may be present in Alzheimer’s disease brains but are difficult to detect using traditional culture-based methods. This approach has been successfully employed in other diseases such as sepsis and pneumonia to identify novel bacterial and viral species, which could also be applicable to Alzheimer’s disease.
Single-cell sequencing can also provide a high-resolution characterization of the microbial communities present in Alzheimer’s disease brains, enabling the identification of specific microbial species or strains that are associated with the disease and elucidating their functional roles within the community.
Single-cell sequencing can shed light on the interactions between microorganisms and host cells in Alzheimer’s disease brains, offering insights into the mechanisms by which microorganisms contribute to disease pathology and identifying potential targets for therapeutic intervention.
The ethical considerations associated with single-cell sequencing (SCS) technology in the context of Alzheimer’s disease research involve several key aspects. Researchers need to prioritize informed consent, ensuring that individuals contributing biological samples are fully aware of the potential risks and benefits. Rigorous data anonymization is crucial to prevent the identification of participants and protect their privacy. Robust data security measures, including encryption and access controls, are essential to safeguarding sensitive information. Transparent data-sharing policies should be established, specifying who has access to the data and for what purposes, while community engagement ensures inclusivity and consideration of diverse perspectives. Long-term data governance structures and regular ethics review board approval contribute to ongoing oversight and adherence to ethical standards. Overall, these measures collectively promote the responsible and ethical use of SCS technology in Alzheimer’s disease research [133-135].
1.17. Ethical Considerations Associated with Single-cell Sequencing (SCS) Technology in the Context of Alzheimer’s Disease
The ethical considerations associated with single-cell sequencing (SCS) technology in the context of Alzheimer’s disease research involve several key aspects. Researchers need to prioritize informed consent, ensuring that individuals contributing biological samples are fully aware of the potential risks and benefits. Rigorous data anonymization is crucial to prevent the identification of participants and protect their privacy. Robust data security measures, including encryption and access controls, are essential to safeguarding sensitive information. Transparent data sharing policies should be established, specifying who has access to the data and for what purposes, while community engagement ensures inclusivity and consideration of diverse perspectives. Long-term data governance structures and regular ethics review board approval contribute to ongoing oversight and adherence to ethical standards. Overall, these measures collectively promote the responsible and ethical use of SCS technology in Alzheimer’s disease research [133-135]. Neuroscience research generates extensive brain data that is crucial for advancements and medical solutions. However, managing this data involves navigating diverse jurisdictions, formats, and regulations. The governance of brain data has become imperative, leading to the establishment of diverse structures to uphold data quality, availability, and ethical considerations. Despite the recognition of ethical and legal principles in data governance, a standardized framework for managing brain data remains unclear due to evolving practices and principles. Authors can follow the cited article for more about neuroethics [135-139].
2. CONCLUSION
Alzheimer’s disease (AD) is a neurodegenerative disorder characterized by the buildup of amyloid plaques and neurofibrillary tangles in the brain. Despite extensive research, the causes of AD remain poorly understood. However, recent evidence suggests that microbial infections may contribute to the development and progression of AD. Single-cell sequencing (SCS) technology has emerged as a powerful tool for investigating the relationship between microbial infections and AD at a high resolution. In this review article, we explore the latest research on the role of SCS in studying complex microbial communities in AD. By enabling the identification and characterization of specific microbial species and their interactions with host cells in AD brains, SCS has provided novel insights into the potential involvement of microbial infections in AD pathogenesis. In addition, SCS has the potential to advance the diagnosis and treatment of AD by uncovering new therapeutic targets. Nevertheless, the challenges, limitations, and ethical and translational considerations of SCS technology must be addressed to ensure its full potential is realized. In conclusion, SCS has significant potential in deepening our understanding of the complex microbial communities in AD and their potential involvement in disease pathogenesis and could ultimately lead to the development of new therapies and preventive measures for this devastating disease.
3. AUTHOR CONTRIBUTIONS
P.S., M.K., and SKG worked tirelessly to write the review article, while M.K. and SKG created the figures that accompanied it. A.C. provided the initial idea for the article and guided the writing process. All authors have read and agreed to the published version of the manuscript.
4. CONFLICTS OF INTEREST
The authors report no financial or any other conflicts of interest in this work.
5. FINANCIAL SUPPORT
We declare that this review paper received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
6. ETHICAL APPROVALS
This study does not involve experiments on animals or human subjects.
7. DATA AVAILABILITY
All the data is available with the authors and shall be provided upon request.
8. USE OF ARTIFICIAL INTELLIGENCE (AI)-ASSISTED TECHNOLOGY
The authors declares that they have not used artificial intelligence (AI)-tools for writing and editing of the manuscript, and no images were manipulated using AI.
9. Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of the publisher, the editors and the reviewers. This journal remains neutral with regard to jurisdictional claims in published institutional affiliation.
REFERENCES
1. Dehhaghi M, Panahi HK, Guillemin GJ. Microorganisms'footprint in neurodegenerative diseases. Front Cell Neurosci 2018;12:466. [CrossRef]
2. Bulgart HR, Neczypor EW, Wold LE, Mackos AR. Microbial involvement in Alzheimer disease development and progression. Mol Neurodegener 2020;15:42. [CrossRef]
3. Lotz SK, Blackhurst BM, Reagin KL, Funk KE. Microbial infections are a risk factor for neurodegenerative diseases. Front Cell Neurosci 2021;15:691136. [CrossRef]
4. Zhang Z, Liu D, Liu S, Zhang S, Pan Y. The role of Porphyromonas gingivalis outer membrane vesicles in periodontal disease and related systemic diseases. Front Cell Infect Microbiol 2021;10:585917. [CrossRef]
5. Itzhaki RF. Herpes simplex virus type 1 and Alzheimer's disease:Increasing evidence for a major role of the virus. Front Aging Neurosci 2014;6:202. [CrossRef]
6. What is Alzheimer's Disease?Symptoms and Causes. Available from:https://www.alz.org/alzheimers-dementia/what-is-alzheimers [Last accessed on 2023 May 01].
7. Dementia. (n.d.). Available from:https://www.who.int/news-room/fact-sheets/detail/dementia [Last accessed on 2023 May 01].
8. Available from:https://www.alz.org/media/homeoffice/facts and figures/facts-and-figures.pdf [Last accessed on 2020 Oct 10].
9. Itzhaki RF, Lathe R, Balin BJ, Ball MJ, Bearer EL, Braak H, et al. Microbes and Alzheimer's disease. J. Alzheimers Dis 2016;51:979-84. [CrossRef]
10. Piekut T, Hurla M, Banaszek N, Szejn P, Dorszewska J, Kozubski W, et al. Infectious agents and Alzheimer's disease. J Integr Neurosci 2022;21:73. [CrossRef]
11. Ou YN, Zhu JX, Hou XH, Shen XN, Xu W, Dong Q, et al. Associations of infectious agents with Alzheimer's disease:A systematic review and meta-analysis. J Alzheimers Dis 2020;75:299-309. [CrossRef]
12. Chaudhary A, Maurya PK, Yadav BS, Singh S, Mani A. Current therapeutic targets for Alzheimer's disease. J Biomed 2018;3:74-84. [CrossRef]
13. Friesner RA, Murphy RB, Repasky MP, Frye LL, Greenwood JR, Halgren TA, et al. Extra precision glide:Docking and scoring incorporating a model of hydrophobic enclosure for protein- ligand complexes. J Med Chem 2006;49:6177-96. [CrossRef]
14. Baulch JE, Acharya MM, Agrawal S, Apodaca LA, Monteiro C, Agrawal A. Immune and inflammatory determinants underlying Alzheimer's disease pathology. J Neuroimmune Pharmacol 2020;15:852-62. [CrossRef]
15. Murphy MP, Levine H. Alzheimer's disease and the b-amyloid peptide. J Alzheimers Dis 2010;19:311. [CrossRef]
16. Hippius H, Neundörfer G. The discovery of Alzheimer's disease. Dialogues Clin Neurosci 2003;5:101-8. [CrossRef]
17. Dominy SS, Lynch C, Ermini F, Benedyk M, Marczyk A, Konradi A, et al. Porphyromonas gingivalis in Alzheimer's disease brains:Evidence for disease causation and treatment with small-molecule inhibitors. Sci Adv 2019;5:eaau3333. [CrossRef]
18. Jensen A, Ladegaard Grønkjær L, Holmstrup P, Vilstrup H, Kilian M. Unique subgingival microbiota associated with periodontitis in cirrhosis patients. Sci Rep 2018;8:10718. [CrossRef]
19. Chacko A, Delbaz A, Walkden H, Basu S, Armitage CW, Eindorf T, et al. Chlamydia pneumoniae can infect the central nervous system via the olfactory and trigeminal nerves and contributes to Alzheimer's disease risk. Sci Rep 2022;12:2759. [CrossRef]
20. Miklossy J. Chronic inflammation and amyloidogenesis in Alzheimer's disease--role of Spirochetes. J Alzheimers Dis 2008;13:381-91. [CrossRef]
21. Olsen I, Singhrao SK. Inflammasome involvement in Alzheimer's disease. J Alzheimers Dis 2016;54:45-53. [CrossRef]
22. Alonso R, Pisa D, Marina AI, Morato E, Rábano A, Carrasco L. Fungal infection in patients with Alzheimer's disease. J Alzheimers Dis 2014;41:301-11. [CrossRef]
23. Laugisch O, Johnen A, Buergin W, Eick S, Ehmke B, Duning T, et al. Oral and periodontal health in patients with Alzheimer's disease and other forms of dementia - a cross-sectional pilot study. Oral Health Prev Dent 2021;19:255-61.
24. Kim SR, Son M, Kim YR, Kang HK. Risk of dementia according to the severity of chronic periodontitis in Korea:A nationwide retrospective cohort study. Epidemiol Health 2022;44:e2022077. [CrossRef]
25. Soreq L, Bird H, Mohamed W, Hardy J. Single-cell RNA sequencing analysis of human Alzheimer's disease brain samples reveals neuronal and glial specific cells differential expression. PLoS One 2023;18:e0277630. [CrossRef]
26. Emery DC, Shoemark DK, Batstone TE, Waterfall CM, Coghill JA, Cerajewska TL, et al. 16S rRNA next generation sequencing analysis shows bacteria in Alzheimer's post-mortem brain. Front Aging Neurosci 2017;9:195. [CrossRef]
27. Li W, Lee MH, Henderson L, Tyagi R, Bachani M, Steiner J, et al. Human endogenous retrovirus-K contributes to motor neuron disease. Sci Transl Med 2015;7:307ra153. [CrossRef]
28. Weaver DF. Alzheimer's disease as an innate autoimmune disease (AD2):A new molecular paradigm. Alzheimers Dement 2023;19:1086-98. [CrossRef]
29. Soscia SJ, Kirby JE, Washicosky KJ, Tucker SM, Ingelsson M, Hyman B, et al. The Alzheimer's disease-associated amyloid beta-protein is an antimicrobial peptide. PLoS One 2010;5:e9505. [CrossRef]
30. Allen HB. Alzheimer's disease:Assessing the role of spirochetes, biofilms, the immune system, and amyloid-b with regard to potential treatment and prevention. J Alzheimers Dis 2016;53:1271-6. [CrossRef]
31. Liu SY, Aliyari R, Chikere K, Li G, Marsden MD, Smith JK, et al. Interferon-inducible cholesterol-25-hydroxylase broadly inhibits viral entry by production of 25-hydroxycholesterol. Immunity 2013;38:92-105. [CrossRef]
32. Blanc M, Hsieh WY, Robertson KA, Kropp KA, Forster T, Shui G, et al. The transcription factor STAT-1 couples macrophage synthesis of 25-hydroxycholesterol to the interferon antiviral response. Immunity 2013;38:106-18. [CrossRef]
33. Papassotiropoulos A, Lambert JC, Wavrant-De Vrièze F, Wollmer MA, Von Der Kammer H, Streffer JR, et al. Cholesterol 25-hydroxylase on chromosome 10q is a susceptibility gene for sporadic Alzheimer's disease. Neurodegener Dis 2005;2:233-41. [CrossRef]
34. White MR, Kandel R, Tripathi S, Condon D, Qi L, Taubenberger J, et al. Alzheimer's associated b-amyloid protein inhibits influenza a virus and modulates viral interactions with phagocytes. PLoS One 2014;9:e101364. [CrossRef]
35. Bourgade K, Le Page A, Bocti C, Witkowski JM, Dupuis G, Frost EH, et al. Protective effect of amyloid-b peptides against herpes simplex virus-1 infection in a neuronal cell culture model. J Alzheimers Dis 2016;50:1227-41. [CrossRef]
36. Bourgade K, Garneau H, Giroux G, Le Page AY, Bocti C, Dupuis G, et al. b-amyloid peptides display protective activity against the human Alzheimer's disease-associated herpes simplex virus-1. Biogerontology 2015;16:85-98. [CrossRef]
37. Williams WM, Torres S, Siedlak SL, Castellani RJ, Perry G, Smith MA, et al. Antimicrobial peptide b-defensin-1 expression is upregulated in Alzheimer's brain. J Neuroinflammation 2013;10:127. [CrossRef]
38. Hwang B, Lee JH, Bang D. Single-cell RNA sequencing technologies and bioinformatics pipelines. Exp Mol Med 2018;50:1-14. [CrossRef]
39. Evrony GD, Hinch AG, Luo C. Applications of single-cell DNA sequencing. Annu Rev Genomics Hum Genet 2021;22:171-97. [CrossRef]
40. Deng Y, Bartosovic M, Ma S, Zhang D, Kukanja P, Xiao Y, et al. Spatial profiling of chromatin accessibility in mouse and human tissues. Nature 2022;609:375-83. [CrossRef]
41. Stuart T, Satija R. Integrative single-cell analysis. Nat Rev Genet 2019;205:257-72. [CrossRef]
42. Van den Berge K, Hembach KM, Soneson C, Tiberi S, Clement L, Love MI, et al. RNA sequencing data:Hitchhiker's guide to expression analysis. Annu Rev Biomed Data Sci 2019;2:139-73. [CrossRef]
43. Patro R, Duggal G, Love MI, Irizarry RA, Kingsford C. Salmon provides fast and bias-aware quantification of transcript expression. Nat Methods 2017;14:417-9. [CrossRef]
44. Hao Y, Stuart T, Kowalski M, Choudhary S, Hoffman P, Hartman A, et al. Dictionary learning for integrative, multimodal, and scalable single-cell analysis. Nat Biotechnol 2023:1-12. [CrossRef]
45. Zhang X, Lan Y, Xu J, Quan F, Zhao E, Deng C, et al. CellMarker:A manually curated resource of cell markers in human and mouse. Nucleic Acids Res 2019;47:D721-8. [CrossRef]
46. Guo H, Zhu P, Yan L, Li R, Hu B, Lian Y, et al. The DNA methylation landscape of human early embryos. Nature 2014;511:606-10. [CrossRef]
47. Zhu S, Qing T, Zheng Y, Jin L, Shi L. Advances in single-cell RNA sequencing and its applications in cancer research. Oncotarget 2017;8:53763-79. [CrossRef]
48. Pan D, Jia D. Application of single-cell multi-omics in dissecting cancer cell plasticity and tumor heterogeneity. Front Mol Biosci 2021;8:757024. [CrossRef]
49. Casado-Pelaez M, Bueno-Costa A, Esteller M. Single cell cancer epigenetics. Trends Cancer 2022;8:820-38. [CrossRef]
50. Lyu T, Lin Y, Wu K, Cao Z, Zhang Q, Zheng J. Single-cell sequencing technologies in bladder cancer research:Applications and challenges. Front Genet 2022;13:1027909. [CrossRef]
51. Heemels MT. Neurodegenerative diseases. Nature 2016;539:179. [CrossRef]
52. Assal F. History of dementia. Front Neurol Neurosci 2019;44:118-26. [CrossRef]
53. Shnayder M, Nachshon A, Rozman B, Bernstein B, Lavi M, Fein N, et al. Single cell analysis reveals human cytomegalovirus drives latently infected cells towards an anergic-like monocyte state. Elife 2020;9:e52168. [CrossRef]
54. Bao J, Li L, Zhang Y, Wang M, Chen F, Ge S, et al. Periodontitis may induce gut microbiota dysbiosis via salivary microbiota. Int J Oral Sci 2022;141:32. [CrossRef]
55. Jungbauer G, Stähli A, Zhu X, Auber Alberi L, Sculean A, Eick S. Periodontal microorganisms and Alzheimer disease - a causative relationship?Periodontol 2000 2022;89:59-82. [CrossRef]
56. Alsema AM, Jiang Q, Kracht L, Gerrits E, Dubbelaar ML, Miedema A, et al. Profiling microglia from Alzheimer's disease donors and non-demented elderly in acute human postmortem cortical tissue. Front Mol Neurosci 2020;13:134. [CrossRef]
57. Wang H. Microglia heterogeneity in Alzheimer's disease:Insights from single-cell technologies. Front Synaptic Neurosci 2021;13:68. [CrossRef]
58. Pettas S, Karagianni K, Kanata E, Chatziefstathiou A, Christoudia N, Xanthopoulos K, et al. Profiling microglia through single-Cell RNA sequencing over the course of development, aging, and disease. Cells 2022;11:2383. [CrossRef]
59. Roy ER, Cao W. Antiviral immune response in Alzheimer's disease:Connecting the dots. Front Neurosci 2020;14:577744. [CrossRef]
60. Chandra S, Sisodia SS, Vassar RJ. The gut microbiome in Alzheimer's disease:What we know and what remains to be explored. Mol Neurodegener 2023;18:9. [CrossRef]
61. Vogt NM, Kerby RL, Dill-McFarland KA, Harding SJ, Merluzzi AP, Johnson SC, et al. Gut microbiome alterations in Alzheimer's disease. Sci Rep 2017;7:13537. [CrossRef]
62. Lin R, Zhou Y, Yan T, Wang R, Li H, Wu Z, et al. Directed evolution of adeno-associated virus for efficient gene delivery to microglia. Nat Methods 2022;198:976-85. [CrossRef]
63. Jiang C, Li G, Huang P, Liu Z, Zhao B. The gut microbiota and Alzheimer's disease. J Alzheimers Dis 2017;58:1-15. [CrossRef]
64. Li B, He Y, Ma J, Huang P, Du J, Cao L, et al. Mild cognitive impairment has similar alterations as Alzheimer's disease in gut microbiota. Alzheimers Dement 2019;15:1357-66. [CrossRef]
65. Liu F, Liu A, Lu X, Zhang Z, Xue Y, Xu J, et al. Dysbiosis signatures of the microbial profile in tissue from bladder cancer. Cancer Med 2019;8:6904-14. [CrossRef]
66. Schirmer M, Franzosa EA, Lloyd-Price J, McIver LJ, Schwager R, Poon TW, et al. Dynamics of metatranscription in the inflammatory bowel disease gut microbiome. Nat Microbiol 2017;33:337-46. [CrossRef]
67. Patel S, Howard D, Chowdhury N, Derieux C, Wellslager B, Yilmaz O, et al. Characterization of human genes modulated by Porphyromonas gingivalis highlights the ribosome, hypothalamus, and cholinergic neurons. Front Immunol 2021;12:646259. [CrossRef]
68. Hu B, Xu P, Ma L, Chen D, Wang J, Dai X, et al. One cell at a time:Droplet-based microbial cultivation, screening and sequencing. Mar Life Sci Technol 2021;3:169-88. [CrossRef]
69. Brase L, You SF, D'Oliveira Albanus R, Del-Aguila JL, Dai Y, Novotny BC, et al. Single-nucleus RNA-sequencing of autosomal dominant Alzheimer disease and risk variant carriers. Nat Commun 2023;14:2314. [CrossRef]
70. Durazzi F, Sala C, Castellani G, Manfreda G, Remondini D, De Cesare A. Comparison between 16S rRNA and shotgun sequencing data for the taxonomic characterization of the gut microbiota. Sci Rep 2021;111:3030. [CrossRef]
71. Lloyd-Price J, Arze C, Ananthakrishnan AN, Schirmer M, Avila-Pacheco J, Poon TW, et al. Multi-omics of the gut microbial ecosystem in inflammatory bowel diseases. Nature 2019;569:655-62. [CrossRef]
72. Truong DT, Franzosa EA, Tickle TL, Scholz M, Weingart G, Pasolli E, et al. MetaPhlAn2 for enhanced metagenomic taxonomic profiling. Nat Methods 2015;12:902-3. [CrossRef]
73. Wood DE, Salzberg SL. Kraken:Ultrafast metagenomic sequence classification using exact alignments. Genome Biol 2014;15:R46. [CrossRef]
74. Rieder F, Mukherjee PK, Pranab Mukherjee CK. Skin microbiome in atopic dermatitis:New insights from clinical trials. J Eur Acad Dermatol Venereol 2023;37:649-50. [CrossRef]
75. Gryaznova M, Lebedeva O, Kozarenko O, Smirnova Y, Burakova I, Syromyatnikov M, et al. Lower genital tract microbiome in early pregnancy in the Eastern European population. Microorganisms 2022;10:2368. [CrossRef]
76. Arceneaux D, Chen Z, Simmons AJ, Campbell JD, Liu Q, Lau KS. A contamination focused approach for optimizing the single-cell RNA-seq experiment. iScience 2023;29:107242. [CrossRef]
77. Slyper M, Porter CB, Ashenberg O, Waldman J, Drokhlyansky E, Wakiro I, et al. A single-cell and single-nucleus RNA-Seq toolbox for fresh and frozen human tumors. Nat Med 2020;265:792-802. [CrossRef]
78. Venema WT, Ramírez-Sánchez AD, Bigaeva E, Withoff S, Jonkers I, Mcintyre RE, et al. Gut mucosa dissociation protocols influence cell type proportions and single-cell gene expression levels. Nature 2022;12:9897. [CrossRef]
79. Vachon PH. Methods for assessing apoptosis and anoikis in normal intestine/colon and colorectal cancer. Methods Mol Biol 2018;1765:99-137. [CrossRef]
80. Lacar B, Linker SB, Jaeger BN, Krishnaswami SR, Barron JJ, Kelder MJ, et al. Nuclear RNA-seq of single neurons reveals molecular signatures of activation. Nat Commun 2016;7:11022. [CrossRef]
81. Yang S, Corbett SE, Koga Y, Wang Z, Johnson E, Yajima M, et al. Decontamination of ambient RNA in single-cell RNA-seq with DecontX. Genome Biol 2020;21:57. [CrossRef]
82. Young MD, Behjati S. SoupX removes ambient RNA contamination from droplet-based single-cell RNA sequencing data. Gigascience 2020;9:giaa151. [CrossRef]
83. Wolock SL, Lopez R, Klein AM. Scrublet:Computational identification of cell doublets in single-cell transcriptomic data. Cell Syst 2019;8:281-91.e9. [CrossRef]
84. Salim A, Molania R, Wang J, Livera AD, Thijssen R, Speed TP. RUV-III-NB:Normalization of single cell RNA-seq data. Nucleic Acids Res 2022;50:e96. [CrossRef]
85. McGinnis CS, Murrow LM, Gartner ZJ. DoubletFinder:Doublet detection in single-cell RNA sequencing data using artificial nearest neighbors. Cell Syst 2019;8:329-37.e4. [CrossRef]
86. Xi NM, Li JJ. Benchmarking computational doublet-detection methods for single-cell RNA sequencing data. Cell Syst 2021;12:176-94.e6. [CrossRef]
87. Huang D, Ma N, Li X, Gou Y, Duan Y, Liu B, et al. Advances in single-cell RNA sequencing and its applications in cancer research. J Hematol Oncol 2023;16:98. [CrossRef]
88. Chen G, Ning B, Shi T. Single-cell RNA-seq technologies and related computational data analysis. Front Genet 2019;10:317. [CrossRef]
89. Haque A, Engel J, Teichmann SA, Lönnberg T. A practical guide to single-cell RNA-sequencing for biomedical research and clinical applications. Genome Med 2017;9:75. [CrossRef]
90. Cuomo AS, Seaton DD, McCarthy DJ, Martinez I, Bonder MJ, Garcia-Bernardo J, et al. Single-cell RNA-sequencing of differentiating iPS cells reveals dynamic genetic effects on gene expression. Nat Commun 2020;11:1. [CrossRef]
91. Rogers GB, Carroll MP, Serisier DJ, Hockey PM, Jones G, Bruce KD. Characterization of bacterial community diversity in cystic fibrosis lung infections by use of 16s ribosomal DNA terminal restriction fragment length polymorphism profiling. J Clin Microbiol 2004;42:5176-83. [CrossRef]
92. Sam LT, Lipson D, Raz T, Cao X, Thompson J, Milos PM, et al. A comparison of single molecule and amplification based sequencing of cancer transcriptomes. PLoS One 2011;6:e17305. [CrossRef]
93. Yuan GC, Cai L, Elowitz M, Enver T, Fan G, Guo G, et al. Challenges and emerging directions in single-cell analysis. Genome Biol 2017;18:84. [CrossRef]
94. McCarthy DJ, Campbell KR, Lun AT, Wills QF. Scater:Pre-processing, quality control, normalization and visualization of single-cell RNA-seq data in R. Bioinformatics 2017;33:1179. [CrossRef]
95. Lun AT, Riesenfeld S, Andrews T, Dao TP, Gomes T, Marioni JC. EmptyDrops:Distinguishing cells from empty droplets in droplet-based single-cell RNA sequencing data. Genome Biol 2019;20:63. [CrossRef]
96. Andrews TS, Kiselev VY, McCarthy D, Hemberg M. Tutorial:Guidelines for the computational analysis of single-cell RNA sequencing data. Nat Protoc 2021;16:1-9. [CrossRef]
97. Muskovic W, Powell JE. DropletQC:Improved identification of empty droplets and damaged cells in single-cell RNA-seq data. Genome Biol 2021;22:329. [CrossRef]
98. Smith-Roe SL, Hobbs CA, Hull V, Auman JT, Recio L, Streicker MA, et al. Adopting duplex sequencing™ technology for genetic toxicity testing:A proof-of-concept mutagenesis experiment with N-Ethyl-N-nitrosourea (ENU)-exposed rats. BioRxiv 2023;891:1-22. [CrossRef]
99. Smith T, Heger A, Sudbery I. UMI-tools:Modeling sequencing errors in unique molecular Identifiers to improve quantification accuracy. Genome Res 2017;27:491-9. [CrossRef]
100. Shestak AG, Bukaeva AA, Saber S, Zaklyazminskaya EV. Allelic dropout is a common phenomenon that reduces the diagnostic yield of PCR-based sequencing of targeted gene panels. Front Genet 2021;12:620337. [CrossRef]
101. Baker SC, Bauer SR, Beyer RP, Brenton JD, Bromley B, Burrill J, et al. The external RNA controls consortium:A progress report. Nat Methods 2005;2:731-4. [CrossRef]
102. Reid LH. Proposed methods for testing and selecting the ERCC external RNA controls. BMC Genomics 2005;6:150. [CrossRef]
103. Waise S, Parker R, Rose-Zerilli MJ, Layfield DM, Wood O, West J, et al. An optimised tissue disaggregation and data processing pipeline for characterising fibroblast phenotypes using single-cell RNA sequencing. Sci Rep 2019;9:9580. [CrossRef]
104. Van Hove H, Martens L, De Vlaminck K, Antunes AR, De Prijck S, Vandamme N, et al. A single-cell atlas of mouse brain macrophages reveals unique transcriptional identities shaped by ontogeny and tissue environment. Nat Neurosci 2019;22:1021-35. [CrossRef]
105. van den Brink SC, Sage F, Vértesy A, Spanjaard B, Peterson-Maduro J, Baron CS, et al. Single-cell sequencing reveals dissociation-induced gene expression in tissue subpopulations. Nat Methods 2017;14:935-6. [CrossRef]
106. Machado L, Geara P, Camps J, Dos Santos M, Teixeira-Clerc F, Van Herck J, et al. Tissue damage induces a conserved stress response that initiates quiescent muscle stem cell activation. Cell Stem Cell 2021;28:1125-35.e7. [CrossRef]
107. Wu YE, Pan L, Zuo Y, Li X, Hong W. Detecting activated cell populations using single-cell RNA-seq. Neuron 2017;96:313-28.e6. [CrossRef]
108. Mattei D, Ivanov A, Van Oostrum M, Pantelyushin S, Richetto J, Mueller F, et al. Enzymatic dissociation induces transcriptional and proteotype bias in brain cell populations. Int J Mol Sci 2020;21:7944. [CrossRef]
109. Qiu P. Embracing the dropouts in single-cell RNA-seq analysis. Nat Commun 2020;111:1169. [CrossRef]
110. AlJanahi AA, Danielsen M, Dunbar CE. An introduction to the analysis of single-cell RNA-sequencing data. Mol Ther Methods Clin Dev 2018;10:189-96. [CrossRef]
111. Kharchenko PV, Silberstein L, Scadden DT. Bayesian approach to single-cell differential expression analysis. Nat Methods 2014;117:740-2. [CrossRef]
112. Svensson V, Natarajan KN, Ly LH, Miragaia RJ, Labalette C, Macaulay IC, et al. Power analysis of single-cell RNA-sequencing experiments. Nat Methods 2017;14:381-7. [CrossRef]
113. Zhang L, Zhang S. Comparison of computational methods for imputing single-cell RNA-sequencing data. IEEE/ACM Trans Comput Biol Bioinform 2020;17:376-89.
114. Tsuyuzaki K, Sato H, Sato K, Nikaido I. Benchmarking principal component analysis for large-scale single-cell RNA-sequencing. Genome Biol 2020;21:9. [CrossRef]
115. Armstrong G, Martino C, Rahman G, Gonzalez A, Vázquez-Baeza Y, Mishne G, et al. Uniform manifold approximation and projection (UMAP) reveals composite patterns and resolves visualization artifacts in microbiome data. mSystems 2021;6:e0069121. [CrossRef]
116. Mulas C, Chaigne A, Smith A, Chalut KJ. Cell state transitions:Definitions and challenges. Development 2021;148:dev199950. [CrossRef]
117. Kujawa T, Marczyk M, Polanska J. Influence of single-cell RNA sequencing data integration on the performance of differential gene expression analysis. Front Genet 2022;13:1009316. [CrossRef]
118. Luecken MD, Theis FJ. Current best practices in single-cell RNA-seq analysis:A tutorial. Mol Syst Biol 2019;15:e8746. [CrossRef]
119. Squair JW, Gautier M, Kathe C, Anderson MA, James ND, Hutson TH, et al. Confronting false discoveries in single-cell differential expression. Nat Commun 2021;121:5692. [CrossRef]
120. Luquez T, Gaur P, Kosater IM, Lam M, Lee DI, Mares J, et al. Cell type-specific changes identified by single-cell transcriptomics in Alzheimer's disease. Genome Med 2022;14:136. [CrossRef]
121. Belonwu SA, Li Y, Bunis D, Rao AA, Solsberg CW, Tang A, et al. Sex-stratified single-cell RNA-Seq analysis identifies sex-specific and cell type-specific transcriptional responses in Alzheimer's disease across two brain regions. Mol Neurobiol 2022;59:276-93. [CrossRef]
122. Masuda T, Sankowski R, Staszewski O, Böttcher C, Amann L, Sagar, et al. Spatial and temporal heterogeneity of mouse and human microglia at single-cell resolution. Nature 2019;566:388-92. [CrossRef]
123. Geirsdottir L, David E, Keren-Shaul H, Weiner A, Bohlen SC, Neuber J, et al. Cross-species single-cell analysis reveals divergence of the primate microglia program. Cell 2019;179:1609-22.e16. [CrossRef]
124. Sankowski R, Böttcher C, Masuda T, Geirsdottir L, Sagar, Sindram E, et al. Mapping microglia states in the human brain through the integration of high -dimensional techniques. Nat Neurosci 2019;2212:2098-110. [CrossRef]
125. Kolodziejczyk AA, Kim JK, Svensson V, Marioni JC, Teichmann SA. The technology and biology of single-cell RNA sequencing. Mol Cell 2015;58:610-20. [CrossRef]
126. Lee MC, Lopez-Diaz FJ, Khan SY, Tariq MA, Dayn Y, Vaske CJ, et al. Single-cell analyses of transcriptional heterogeneity during drug tolerance transition in cancer cells by RNA sequencing. Proc Natl Acad Sci U S A 2014;111:E4726-35. [CrossRef]
127. Marinov GK, Williams BA, McCue K, Schroth GP, Gertz J, Myers RM, et al. From single-cell to cell-pool transcriptomes:Stochasticity in gene expression and RNA splicing. Genome Res 2014;24:496-510. [CrossRef]
128. Sekiya M, Wang M, Fujisaki N, Sakakibara Y, Quan X, Ehrlich ME, et al. Integrated biology approach reveals molecular and pathological interactions among Alzheimer's Ab42, Tau, TREM2, and TYROBP in Drosophila models. Genome Med 2018;10:26. [CrossRef]
129. Iacono D, Resnick SM, O'Brien R, Zonderman AB, An Y, Pletnikova O, et al. Mild cognitive impairment and asymptomatic Alzheimer disease subjects:Equivalent b-amyloid and tau loads with divergent cognitive outcomes. J Neuropathol Exp Neurol 2014;73:295-304. [CrossRef]
130. Lambert JC, Ibrahim-Verbaas CA, Harold D, Naj AC, Sims R, Bellenguez C, et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer's disease. Nat Genet 2013;45:1452-8. [CrossRef]
131. Kachroo AH, Laurent JM, Yellman CM, Meyer AG, Wilke CO, Marcotte EM. Systematic humanization of yeast genes reveals conserved functions and genetic modularity. Science 2015;348:921-5. [CrossRef]
132. Sahni N, Yi S, Taipale M, Fuxman Bass JI, Coulombe-Huntington J, Yang F, et al. Widespread macromolecular interaction perturbations in human genetic disorders. Cell 2015;161:647-60. [CrossRef]
133. Hyun I, Scharf-Deering JC, Lunshof JE. Ethical issues related to brain organoid research. Brain Res 2020;1732:146653. [CrossRef]
134. Farahany NA, Greely HT, Hyman S, Koch C, Grady C, Pasca SP, et al. The ethics of experimenting with human brain tissue. Nature 2018;556:429. [CrossRef]
135. Presley A, Samsa LA, Dubljevi?V. Media portrayal of ethical and social issues in brain organoid research. Philos Ethics Humanit Med 2022;17:8. [CrossRef]
136. Adams A, Albin S, Amunts K, Asakawa T, Bernard A, Bjaalie JG, et al. International brain initiative:An innovative framework for coordinated global brain research efforts. Neuron 2020;105:212-6. [CrossRef]
137. Amadio J, Bi GQ, Boshears PF, Carter A, Devor A, Doya K, et al. Neuroethics questions to guide ethical research in the international brain initiatives. Neuron 2018;100:19-36. [CrossRef]
138. Illes J, Bird SJ. Neuroethics:A modern context for ethics in neuroscience. Trends Neurosci 2006;29:511-7. [CrossRef]
139. Ochang P, Stahl BC, Eke D. The ethical and legal landscape of brain data governance. PLoS One 2022;17:e0273473. [CrossRef]
140. Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, et al. QIIME allows analysis of high-throughput community sequencing data. Nat Methods 2010;75:335-6. [CrossRef]
141. Meyer F, Paarmann D, D'Souza M, Olson R, Glass EM, Kubal M, et al. The metagenomics RAST server - A public resource for the automatic phylogenetic and functional analysis of metagenomes. BMC Bioinform 2008;9:386. [CrossRef]
142. Beghini F, McIver LJ, Blanco-Míguez A, Dubois L, Asnicar F, Maharjan S, et al. Integrating taxonomic, functional, and strain-level profiling of diverse microbial communities with biobakery 3. Elife 2021;10:e65088. [CrossRef]
143. Kostic AD, Ojesina AI, Pedamallu CS, Jung J, Verhaak RG, Getz G, et al. PathSeq:A comprehensive computational tool for the identification or discovery of microorganisms by deep sequencing of human tissue. Nat Biotechnol 2011;29:393-6. [CrossRef]
144. Riffle M, May DH, Timmins-Schiffman E, Mikan MP, Jaschob D, Noble WS, et al. MetaGOmics:A web-based tool for peptide-centric functional and taxonomic analysis of metaproteomics data. Proteomes 2018;6:2. [CrossRef]
145. Huson DH, Beier S, Flade I, Górska A, El-Hadidi M, Mitra S, et al. MEGAN community edition - interactive exploration and analysis of large-scale microbiome sequencing data. PLoS Comput Biol 2016;12:e1004957. [CrossRef]
146. Langille MG, Zaneveld J, Caporaso JG, McDonald D, Knights D, Reyes JA, et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat Biotechnol 2013;319:814-21. [CrossRef]
147. Kang DD, Froula J, Egan R, Wang Z. MetaBAT, an efficient tool for accurately reconstructing single genomes from complex microbial communities. PeerJ 2015;3:e1165. [CrossRef]
148. Arya J, Sharma D, Kumar D, Jakhar R, Khichi A, Dangi M, et al. Comparative genome analysis of Streptococcus strains to identify virulent genes causing neonatal meningitis. Infect Genet Evol 2023;107:105398. [CrossRef]
149. Bonfili L, Cecarini V, Berardi S, Scarpona S, Suchodolski JS, Nasuti C, et al. Microbiota modulation counteracts Alzheimer's disease progression influencing neuronal proteolysis and gut hormones plasma levels. Sci Rep 2017;71:2426. [CrossRef]
150. Readhead B, Haure-Mirande JV, Funk CC, Richards MA, Shannon P, Haroutunian V, et al. Multiscale analysis of independent Alzheimer's cohorts finds disruption of molecular, genetic, and clinical networks by human herpesvirus. Neuron 2018;99:64-82.e7. [CrossRef]
151. Zhan X, Stamova B, Jin LW, Decarli C, Phinney B, Sharp FR. Gram-negative bacterial molecules associate with Alzheimer disease pathology. Neurology 2016;87:2324-32. [CrossRef]