
ARTICLE HIGHLIGHTS
- Candidate nine common miRNAs targeted four ALS-hr-Gs.
- SOD1 and FUS gene expression may inhibit by common miRNAs (miR-3163 and miR-4422) at post-transcriptional.
- SOD1 gene upregulated by three TFs (CEBPD, EGR1, and SP1) and FUS downregulated by TBP.
- Regulatory 3-node motif analysis showed two TFs (TBP and EGR1) upregulated miR-4422 and four TFs (CEBPD, EGR1, SP1, and TBP) upregulated miR-3163.
- Candidate nine miRNAs enriched with neuron functions, miR-422 is highly expressed in brain as compared to miR-3163.
- C9ORF72, TARDBP, FUS majorly expressed in cerebellar hemisphere region of brain but SOD1, highly expressed in frontal cortex-region.
- Spatiotemporal expression analysis showed ALS-hr-Gs expressed in different areas of brain during life span.
1. INTRODUCTION
Amyotrophic lateral sclerosis (ALS) is a rare and fatal neurological disorder affects motor neuron [1]. Globally, the rate of prevalence observed in adulthood is 5.4/100,000 [2]. In patients, two types were found familial ALS (fALS) and sporadic ALS (sALS) [3]. Over the past year, significant research helps to identifying number of genes related to fALS and sALS [4]. Approximately 70% of fALS cases are caused by the most commonly altered genes; superoxide dismutase 1 (SOD1), chromosome 9 open reading frame 72 (C9ORF72), TAR DNA-binding protein 43 (TARDBP), and fused in sarcoma (FUS) [5-8]. Despite this, fALS accounts for about 10% of reported cases, whereas the genetic basis of sALS is largely unclear [4], nevertheless C9ORF72 is well-known, it only contributes around 5% of sALS cases [9]. The association percentage of genes (C9ORF72, FUS, SOD1, and TARDBP) reported in previous studies shown in Table 1 [10]. Due to multigenic nature of the disease, the genetic structure of ALS is complex and its associated gene mutations frequency varies. It implies that ALS has a significant amount of missing heredity to understand it pathogenesis. In the past years, clinical and computational investigations have aided in providing extensive knowledge of the disease-causing genes and their variations in ALS. Various pathophysiological pathways have been reported that disrupt and lead to ALS pathogenesis such as RNA metabolism, protein homeostasis, DNA repair mechanisms, damage of nucleocytoplasmic transport, excitotoxicity, mitochondrial and axonal transport disturbance, oxidative stress, dysfunction of oligodendrocyte, and vesicular transport aberrations [10-12]. On the basis of ALS-genes associated pathophysiology with other neurodegenerative disease such as frontotemporal dementia, progressive muscular atrophy, primary lateral sclerosis, and hereditary spastic paraplegia, ranking of four genes (C9ORF72, FUS, SOD1, and TARDBP) were done [Figure 1] [13-15]. Four genes C9ORF72, FUS, SOD1, and TARDB have been found to cause ALS pathogenesis and were considered ALS high-risk genes (ALS-hr-Gs) [Figure 1]. Other than four ALS has been linked to a large number of genes, these genes might not always be harmful, and specific mutations in recognized disease-causing genes might not always be pathogenic [15]. Consequently, it is rarely simple to link a genetic alteration to the ALS clinical phenotype. Therefore, understanding the pathogenic impact of ALS associated mutations is necessarily required to explain the disease-associated pathophysiology.
Table 1: Association percentage of high-risk genes in amyotrophic lateral sclerosis.
| Gene | Disease Subtype (OMIM) | Inheritance | Chromos-omal locus | Freque-ncy (%) | fALS % | sALS % | Onset phenotype | Phenotypes | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Spinal Onset (%) | Bulbar onset (%) | UMN (%) | LMN (%) | UMN+LMN (%) | FTD (%) | |||||||
| C9ORF72 | ALS-FTLD 2 and FTD-ALS 1 (614260) | AD | 9p21.2 | ~8 | 35 | 7 | 33.10 | 39.00 | 17.70 | 13.50 | 72.70 | 35.60 |
| SOD1 | ALS1 (147450) | AD, AR | 21q22.11 | ~3 | 15 | 2 | 95.00 | 1.70 | 7.10 | 47.60 | 45.20 | 0.00 |
| FUS | ALS6 (137070) | AD | 16p11.2 | ~0.9–1.2 | 4 | 1 | 74.20 | 19.60 | 9.50 | 19.00 | 66.70 | 7.90 |
| TARDBP | ALS10 (605078) | AD | 1p36.22 | ~1.2 | 4 | 1 | 44.00 | 23.90 | 3.70 | 18.50 | 44.40 | 12.40 |
AD: Autosomal dominant, AR: Autosomal recessive, ALS: Amyotrophic lateral sclerosis, fALS: Familial ALS, sALS: Sporadic ALS, OMIM: Online mendelian inheritance in man, UMN: Upper motor neuropathies, LMN: Lower motor neuropathies, FTD: Frontotemporal dementia
 | Figure 1: Representation of the complexity of amyotrophic lateral sclerosis (ALS) pathogenesis. The inner circle includes the associated genes with highest frequency (chromosome 9 open reading frame 72, SOD1, TAR DNA-binding protein 43, and fused in sarcoma). The second-order ring consist the large number of genes with a lower frequency of association. The third-order ring consists the possible pathogenic mechanisms that are hypothesized to be associated with inner and second circle genes. The outer most ring consists the overlapped diseases that may be associated with ALS genes (inner and second circle). The complex relationship between genes associated with neurodegeneration, mechanisms of neurodegeneration, and clinical disease phenotypes is apparent. Orange square = major genes; pink rectangle = other ALS genes; green circle = disease pathophysiology; yellow circle = high frequent disease pathophysiology; blue diamond shape = associated diseases. Gene-mechanisms connections are shown by red arrow, and gene-disease association by blue arrow. [Click here to view] |
The expression of gene can be control at the level of transcriptional and post-transcriptional inside the cell to direct the normal function. The two layered regulatory network approaches, in which transcription factors (TFs) function as a mediator for microRNAs (miRNAs) regulatory process helps to elucidate the expression level of genes [16,17]. TFs can activate or repress the transcription process of gene by binding to the specific location in promotor regions and regulate gene expression at transcription level. Other small non-coding RNA molecule miRNA ~22nt in length repress mRNA translation process using mRNA decay or inhibition of mRNA translation and results in altered targeted gene expression at post-transcriptional level [18]. It has been found that miRNAs and mRNA transcription process is regulated by single or multiple TFs and also the expression of mRNA, including TFs, could be controlled by miRNAs [19]. miRNAs involved in various biological functions and diseases, specifically in multifactorial nature diseases, gives an opportunity to understand their mechanisms [20]. To date, we have a better understanding of miRNA biogenesis process, function and their presence in biofluids including urine, blood, and cerebrospinal fluid [21,22]. Various studies have been reported that miRNAs are differentially expressed (up- or downregulation) in ALS patients [23,24]. The tissue-specific expression nature of miRNAs helps to understand to distinguish between normal and disease progression of respective tissue [25-27]. Several studies reported the important role of miRNA molecules in physiological and pathological processes, such as immune functions, metabolic pathways, tumorigenesis, and many neurodegenerative diseases [28-31]. Number of miRNAs such as (miR-451, miR-1275, miR-328, miR-638, miR-149, miR-665, and miR- 338-3p) were predicted as biomarker for ALS in human samples [19]. Many studies reported that miRNAs expression in brain are involved in microglia activation and inflammation processes [32,33]. They also play an important role in synaptic plasticity, neuron death or degeneration, and neuron development [34]. In this research article, we applied a bioinformatics approach to identified potential regulators of four ALS-hr-Gs. A combinatorial ALS-hr-Gs network along their transcriptional (TFs) and post-transcriptional regulators (miRNAs) were constructed. Furthermore, we performed the GO function analysis such as biological process (BP), molecular functions (MFs), and cellular component (CC) suggested that these miRNAs participated in many metabolic activities and biological functions that may play an important role in ALS. We also predicted the association of candidate miRNAs with biological pathways, relation with other diseases and their expression pattern in brain. We also investigated the gene expression pattern of ALS-hr-Gs at different age groups along with different parts of the brain. We were interested in predicting novel molecular regulators at motif level to understanding the pathogenesis of ALS.
2. METHODS
2.1. Candidate miRNA and TF Prediction of ALS-hr-Gs
To identify the miRNA target of ALS-hr-Gs, we used three miRNA target prediction tool TargetScan V8.0 (https://www.targetscan.org/) [35], mirTarBase (https://mirtarbase.cuhk.edu.cn/) [36], and miRDB (http://mirdb.org/) [37] databases. After removing redundancy, we considered only those miRNAs which were commonly present in all of the three databases. From the commonly predicted miRNAs of three databases, we filtered those miRNAs targeting all four ALS-hr-Gs considered candidate miRNAs. The transcriptional regulatory relationships unraveled by sentence-based text-mining (TRRUST) a transcription binding database [38], were used to identify the TFs for ALS-hr-Gs. The analysis of “TRRUST” database between TFs and genes was based on manual curation of Medline abstract and regulatory interactions inferred from high-throughput expression data. On the basis of regulatory function, TFs were categories as (i) activators and (ii) repressors molecules.
2.2. Regulatory Relationship between TFs and miRNAs
To analyze the regulatory relationship between predicted candidate miRNAs and TFs, we took the miRNA and TFs relationship from TransmiR v2.0 database [39]. TransmiR v2.0 database manually curate TFs-miRNA regulatory relationship based on research publications and experimentally derived ChIP-seq-analysis. Here, we downloaded tsv.gz format data file (all evidence level) from database contains TF-miRNA relation in human.
2.3. Construction of a Combinatorial ALS-hr-Gs Network
To construct the combinatorial ALS-hr-Gs network, we used three relationship (i) miRNA-ALS-hr-Gs, (ii) ALS-hr-Gs-TFs, and (iii) miRNAs-TFs. The combinatorial ALS-hr-Gs network was constructed and visualized using Cytoscape v3.8.2 [40]. On the basis of 3-node motif (TF-miRNA-gene) analysis, we identified both FFLs (coherent and in-coherent) in ALS-hr-Gs combinatorial network. We considered the general mechanisms of mRNA translation inhibition showed by miRNAs at post-transcriptional [41,42].
2.4. Functional Annotation of Candidate miRNAs
To understand the biological significance of the candidate miRNAs, we used the same prediction tools and approach implemented in miRNA target prediction for ALS-hr-Gs. We performed the functional annotation of candidate miRNAs in terms of MF, BP and CC using the ShinyGO v0.75 tool (http://bioinformatics.sdstate.edu/go/). Furthermore, we done the pathway analysis of candidate miRNAs [43] using Wikipathways dataset in ShinyGO v0.75 tool. The functional analysis plotted using the fold enrichment statistics represented the percentage of genes in list belonging to a process, divided by the corresponding percentage in the background and false discovery rate used with a cutoff (0.05) indicated how likely the enrichment is by chance.
2.5. Candidate miRNAs-diseases Association
To analyze the association of the candidate miRNAs with other diseases, we used WEB-based GEne SeT AnaLysis Toolkit (WebGestalt) tool [44]. We done the analyses based on over representation analysis enrichment method, enrichment categories (disease_OMIM), ID type (gene symbols), reference list (genome_protein_coding) along the parameters such as FDR (FDR < 0.05) used (Benjamini–Hochberg) method.
2.6. Analysis of Candidate miRNAs Expression Pattern
We utilized the DIANA-miTED (miRNA tissue expression database) [45] to understand the expression pattern of predicted candidate miRNAs targeting all four ALS-hr-Gs. Information about miRNA expression and distribution across cell types and tissues is crucial to the understanding of their function and for their translational use as biomarkers or therapeutic targets. DIANA-miTED is the database consist miRNA expression values obtained from the results of 15,183 raw human small RNA-Seq (sRNA-Seq) datasets from the sequence read archive and the cancer genome atlas. The miRNA expression values were used to make box plot in terms of read counts, reads per million (RPM), and log2 (RPM) [45]. Here, we selected the log2 (RPM) expression value for our data to obtained the relationship between the input tissue (brain) and expression of candidate miRNAs.
2.7. Validation of ALS-hr-Gs Expression Pattern
We used the GTEx platform [46] to validate the expression pattern of ALS-hr-Gs in different regions of the brain. Using violin plot, we represented the expression data on the basis of median value. Using the BEST online server, for brain expression spatiotemporal pattern analysis, we generated the spatiotemporal heat map of ALS-hr-Gs. The inputs were utilized by BEST server (gene list) without P-value, logarithm transformation (N), SNP mapping rule (within gene), gene P-value correction (Bonferroni; 0.05), and reference data (RNAseq data from Brainspan) [47].
2.8. Ranking of Candidate miRNAs and TFs
To identify the miRNA and TF which may highly govern the most crucial risk in generating the pathophysiology of ALS, we used a list of ALS genes targeted by candidate miRNAs and TF and also their presence at 3 nodes motif level (coherent and in-coherent feed-forward loops). We provided, the rank-1 for those miRNA and TF targeted the maximum number of ALS related genes and also regulating ALS-hr-Gs at 3 nodes motif level.
3. RESULTS
3.1. Combinatorial Regulatory Network
The miRNA-targets commonly identified by all three prediction programs for SOD1, C9ORF72, TARDBP, and FUS were 130, 559, 909, and 1180, respectively [Table 2]. From the TRRUST database, we found 9 TFs which regulates (activation/repression) SOD1 and FUS. Using the TransmiR v2.0 database, 167 TFs upregulate the expression of miR-422 and miR-3163. The regulator-target relationships in the combinatorial ALS-hr-Gs network were made up of transcriptional and post-transcriptional regulators which involved 2057 nodes and 3086 edges [Figure 2a and Table 3]. In the combinatorial interaction, it was found that of the 4 ALS-hr-Gs, 2 functioned as TFs. From the combinatorial ALS-hr-Gs network, we found nine miRNAs (miR-1468-3p, miR-3163, miR-33a-3p, miR-3691-3p, miR-4422, miR-4666a-3p, miR-513a-5p, miR-6504-3p, and miR-6835-3p) targets all four ALS-hr-Gs [Figure 2a] and nine TFs (CEBPD, EGR1, KLF4, MSX2, MTF1, NFE2L2, PPARD, SP1, and TBP) regulates two of the ALS-hr-Gs (SOD1 and FUS) [Figure 2a]. SOD1 gene was upregulated by six TFs (CEBPD, EGR1, MSX2, MTF1, NFE2L2, and PPARD) and downregulated by two TFs (KLF4 and SP1), whereas TBP downregulated FUS. At post-transcriptional regulation, three TFs (SP1, TBP, and EGR1) activates two miRNAs (miR-3163 and miR-4422). The miR-3163 and miR-4422 were also commonly targeted by few numbers of TFs [Figure 2a]. The coherent and in-coherent FFL analysis, revealed SOD1 directly activated by TFs (SP1, EGR1, and CEBPD) and also inhibited by SP1 [Figure 2b]. From the network analysis, we concluded the potential regulators of ALS were four TFs (SP1, EGR1, CEBPD, and TBP) target two miRNAs; miR-3163 and miR-4422 and these miRNAs negatively regulate the gene expression of SOD1 and FUS at post-transcriptional level [Figure 3].
Table 2: Number of miRNAs targets predicted by different predictions tools.
| Gene name | Gene Id | Uni prot Id | Target scan | miRDB | miTar base | Total miRNA (After removing redundancy) |
|---|---|---|---|---|---|---|
| SOD1 | 6647 | P00441 | 113 | 26 | 200 | 131 |
| C9ORF72 | 203228 | Q96LT7 | 537 | 236 | 364 | 559 |
| TARDBP | 23435 | Q13148 | 1195 | 191 | 1131 | 909 |
| FUS | 2521 | P35637 | 1939 | 133 | 559 | 1180 |
miRNAs: microRNAs, SOD1: Superoxide dismutase 1, C9ORF72: Chromosome 9 open reading frame 72, TARDBP: TAR DNA-binding protein 43, FUS: Fused in sarcoma
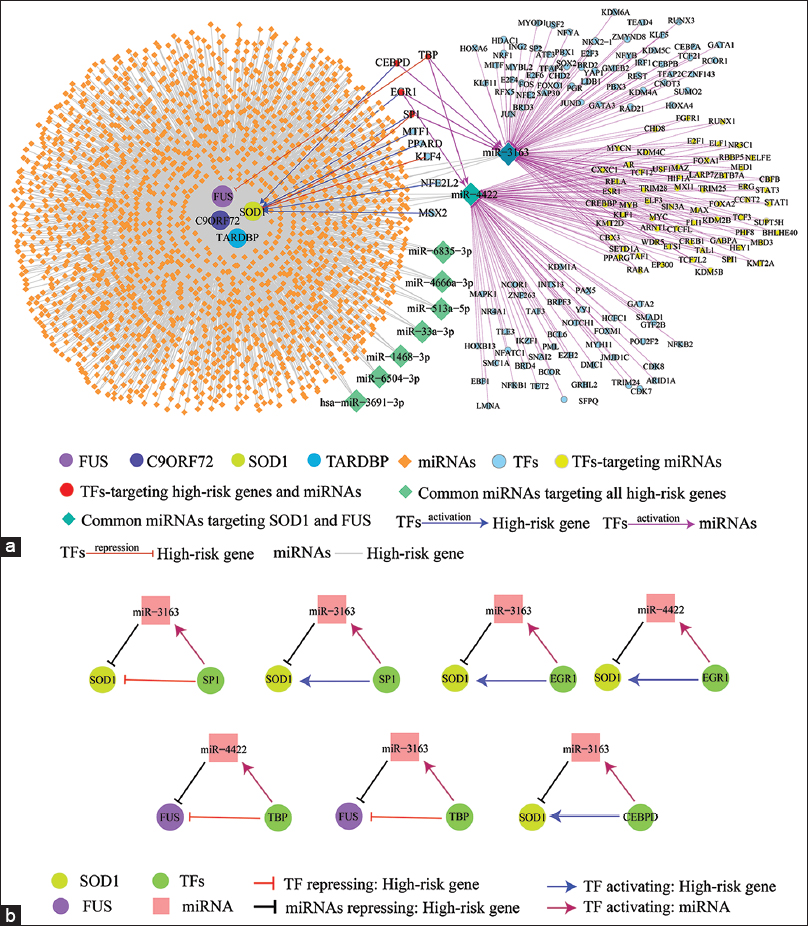 | Figure 2: The combinatorial amyotrophic lateral sclerosis (ALS) high risk gene network (a). The complete gene regulatory network. Nodes: ALS-hr-Gs in different color filled circle, miRNAs represented in orange diamond shape and nine candidate miRNAs showed in light green diamond shape common targets of four ALS-hr-Gs, uncommon transcription factor (TFs) shown in cyan filled circle, common TF targeting miRNA represented in yellow filled circle, common TF targeted both miRNA and ALS-hr-Gs showed in red filled circle. Edges: blue arrow represented (activation; TFs to high-risk genes), Red T-shape edges represented (repression; TFs to high-risk genes), pink arrow represented (activation; TFs to miRNAs), grey line represented (miRNA- ALS-hr-Gs relationship) and (b) Three node motif regulation between TF: miRNA:gene. [Click here to view] |
Table 3: Statistics of nodes and edges in the combinatorial amyotrophic lateral sclerosis-high-risk gene network.
| Nodes/relationship | No. of unique objects |
|---|---|
| Entities of the network | |
| TFs | 174 |
| miRNAs | 1881 |
| Genes (non-TFs) | 2 |
| Total no. of nodes | 2057 |
| Regulatory relationship | |
| TFs–genes | 9 |
| miRNAs–genes | 2844 |
| miRNAs–TFs | 233 |
| Total no. of edges | 3086 |
miRNAs: microRNAs, TFs: Transcription factors
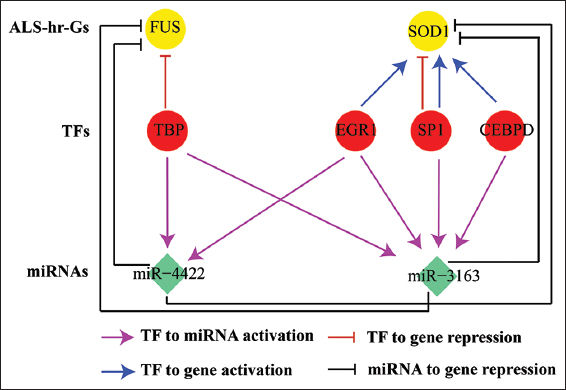 | Figure 3: The potential regulators of amyotrophic lateral sclerosis (ALS) high risk genes. The potential regulators of ALS-hr-Gs SOD1 and FUS showed in (yellow filled circle) are four transcription factor (TFs) (SP1, EGR1, CEBPD and TBP) represented in red filled circle target two miRNAs (miR-3163 and miR-4422) showed in light green filled diamond shape. The mode of regulation from TFs to miRNA showed in pink arrow, TF to gene activation in blue arrow and TFs to gene repression in red T-shape, miRNA to gene repression in black curved T-shape. [Click here to view] |
3.2. Top Rank Candidate miRNAs and TFs
We found, miR-3163 as rank-1, in nine candidate miRNAs (miR-1468-3p, miR-3163, miR-33a-3p, miR-3691-3p, miR-4422, miR-4666a-3p, miR-513a-5p, miR-6504-3p, and miR-6835-3p) targeted other 26 ALS genes (PFN1, TARDBP, UNC13A, ALS2, ERBB4, C9ORF72, PON2, SPG11, SETX, VAPB, TBK1, NEK1, MATR3, TIA1, CHRNA3, CHMP2B, KIFAP3, EPHA4, SOD1, ANG, HNRNPA2B1, GLE1, FIG4, PON3, VCP, and FUS) [Table 4] and also presented at 3-node motif level, regulating the SOD1 and FUS [Figure 2b]. We identified, SP1 as a rank-1 TF out of nine TFs, were targeting 4 ALS genes (SOD1, SQSTM, CHRNB4, and PON1) [Table 5] and also presented at 3-node motif level, regulating the function of SOD1 and rank-1 miRNA (miR-3163) [Figure 3].
Table 4: Ranking of candidate miRNAs.
| RANK | miRNA | Number of ALS gene targeted |
|---|---|---|
| 1 | miR-3163* | 26 |
| 2 | miR-1468-3p | 22 |
| 3 | miR-33a-3p | 20 |
| miR-6504-3p | 20 | |
| 4 | miR-513a-5p | 19 |
| miR-4666a-3p | 19 | |
| 5 | miR-4422 | 17 |
| 6 | miR-3691-3p | 14 |
| 7 | miR-6835-3p | 5 |
This table shows the ranking of candidate nine micro-RNAs (miRNAs). The ranking was done on the basis of number of targeted amyotrophic lateral sclerosis (ALS)-associated genes. The “*” represented the miRNA (miR-3163) showed the highest number of targeted genes were considered as rank-1. The miRNAs showed the same number of ALS genes targets, provided the same ranking
Table 5: Ranking of candidate transcription factors.
| TF | Number of ALS gene-target | Name of ALS gene-target | Ranking |
|---|---|---|---|
| SP1* | 4 | SOD1, SQSTM, CHRNB4, PON1 | 1 |
| PPARD | 2 | SOD1, ANG | 2 |
| CEBPD | 1 | SOD1 | 3 |
| EGR1 | 1 | SOD1 | |
| KLF4 | 1 | SOD1 | |
| MSX2 | 1 | SOD1 | |
| MTF1 | 1 | SOD1 | |
| NFE2L2 | 1 | SOD1 |
SOD1: Superoxide dismutase 1, This table shows the ranking of candidate nine transcription factors (TFs). The ranking was done on the basis of number of targeted amyotrophic lateral sclerosis (ALS)-associated genes. The “#” represented the TF (SP1) showed the highest number of targeted genes were considered as rank-1. The TFs showed the same number of ALS genes targets, were provided the same ranking
3.3. Functional Annotation of Candidate miRNAs
All nine predicted miRNAs targeting ALS-hr-Gs were functionally annotated revealed their functions in BP, MF, and their involvement in the regulation of CC. These candidate miRNAs were mostly biologically associated in neuron differentiation, neurogenesis, and positive regulation of RNA biosynthetic processes [Figure 4a]. The MF enrichment analysis represented their major involvement in DNA binding and transcription cis-regulatory DNA binding [Figure 4b]. The CC results stated that the miRNAs association with different cell component such as glutamatergic synapse, chromosome, neuron projection, and synapse [Figure 4c].
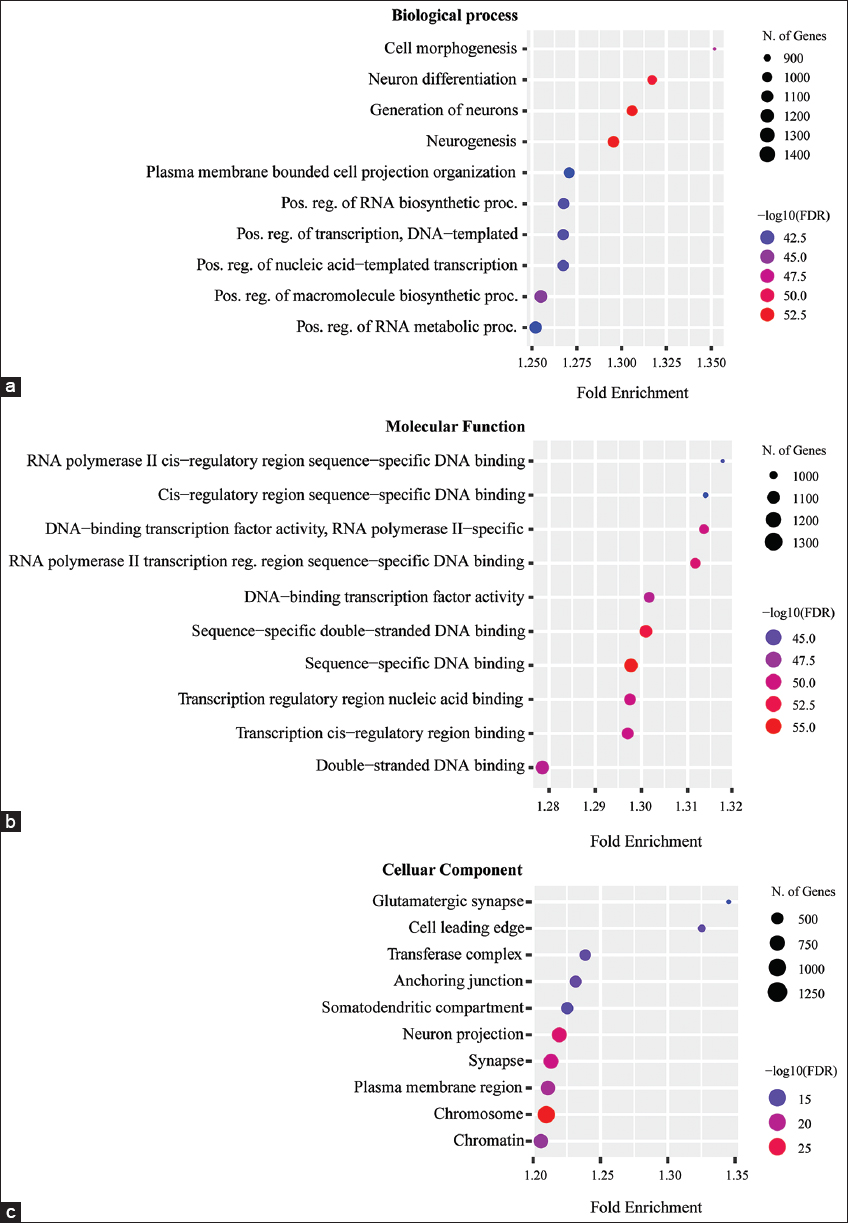 | Figure 4: Functional annotation of candidate miRNAs (a). Representation of biological process (BP) of nine candidate miRNAs in the form of dot plot. The FDR value were calculated on the basis of nominal P-value (0.05) from hygrometric test and fold enrichment represented the percentage of input genes related to a GO term. The size of the circle belongs to the number of genes belongs a particular GO term and (b) Representation of molecular function (MF) of nine candidate miRNAs in the form of dot plot. The FDR value were calculated on the basis of nominal P-value (0.05) from hygrometric test and fold enrichment represented the percentage of input genes related to a GO term. The size of the circle belongs to the number of genes belongs a particular GO term and (c) Representation of CC of nine candidate miRNAs in the form of dot plot. The FDR value were calculated on the basis of nominal P-value (0.05) from hygrometric test and fold enrichment represented the percentage of input genes related to a GO term. The size of the circle belongs to the number of genes belongs a particular GO term. [Click here to view] |
3.4. Pathway, Disease Association, and miRNAs Expression Profiling
The pathways enrichment annotation results showed the nine-candidate miRNA were highly associated with metabolic pathways, pathways in cancer [48-50] and Herpes simplex virus 1 infection [51,52] [Figure 5a]. The disease-association enrichment analysis showed high association with leukemia, breast cancer, diabetes mellitus and tracheoesophageal fistula [Figure 5b]. The miRNA expression pattern revealed two miR-33a-3p and miR-4422 were highly expressed in brain [Figure 5c].
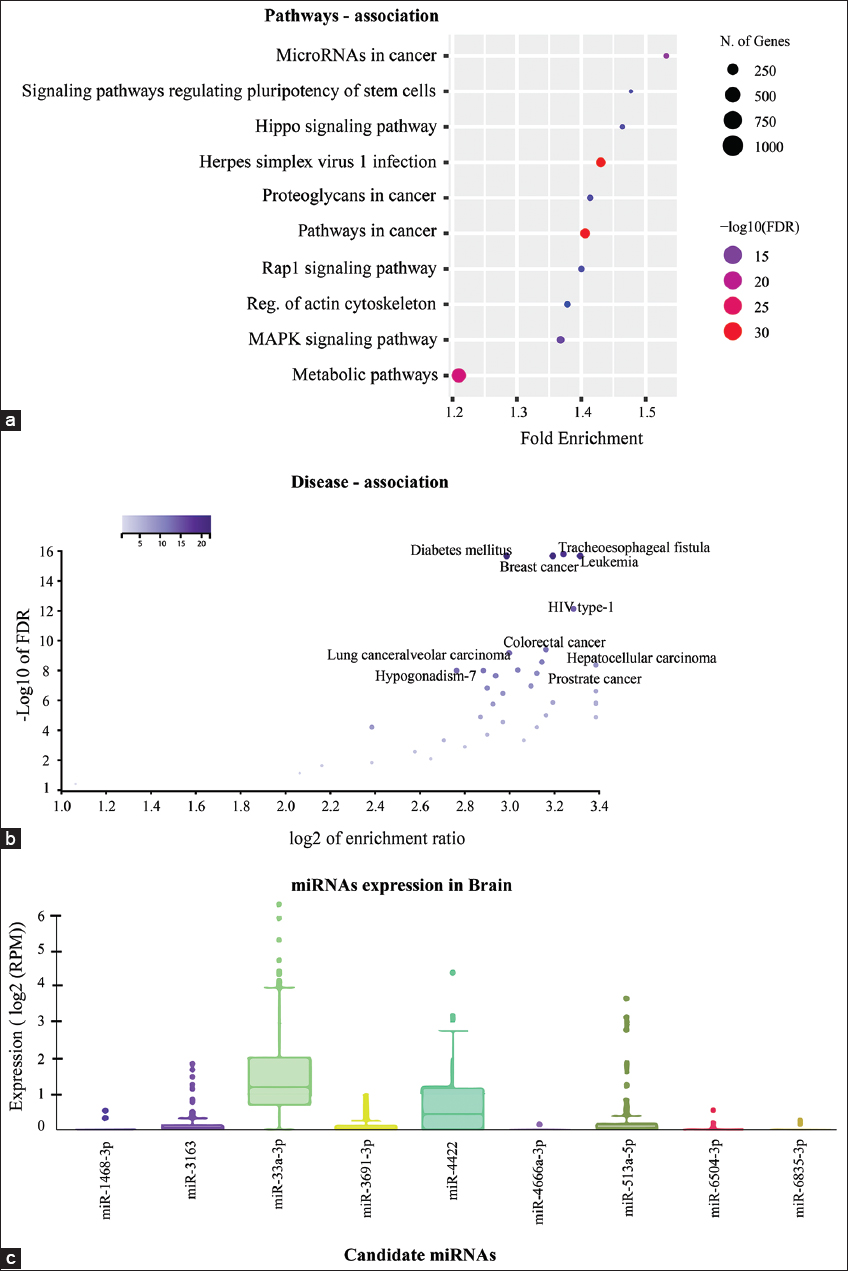 | Figure 5: (a) Representation of the pathway analysis of candidate miRNAs in the form of dot plot. The FDR value were calculated on the basis of nominal P-value (0.05) from hygrometric test and fold enrichment represented the percentage of input genes related to a GO term. The size of the circle belongs to the number of genes belongs a particular GO term. (b) Representation of the possible predicted disease association analysis of nine candidate miRNAs and (c) Expression profiling of candidate miRNAs in brain, different colour box plot represented the expression of a particular miRNA on the basis of log2 (RPM). [Click here to view] |
3.5. Expression Pattern of ALS-hr-Gs
The collected information about the expression pattern of ALS-hr-Gs were analyzed using the violin plot showing the expression pattern in the brain. The violin plot of SOD1 gene expression [Figure 6a], in terms of transcript per million (TPM) is highest, especially in the frontal cortex (BA9) region, and the TPM value of other genes showed (C9ORF72, TARDBP, and FUS) highly expressed in cerebellar hemisphere part of the brain [Figure 6b-d]. Both SOD1 and C9ORF72, showed less expression in basal ganglia region (putamen) but TARDBP, FUS showed less expression in anterior cingulate cortex (BA24) and amygdala region. Spatiotemporal expression analysis showed different areas of the brain like frontal cortex, parietal cortex, temporal cortex, occipital cortex, hippocampus, and cerebellum in mainly involved in expression of ALS-hr-Gs from middle adulthood (40 Y < = age <60 Y) to early fetal 8 (PCW < = age < 13 PCW) age. Furthermore, we also observed a continuous expression of genes [Figure 6e] in most of the areas of the brain at different ages of the life cycle. Some regions of the brain (insula, parahippocampal gyrus, substantia nigra, nucleus accumbens, olfactory bulb, and hypothalamus) have not seen any gene expression at any age of the life span. Both the results showed high and continuous expression of ALS-hr-Gs in frontal cortex and cerebellum parts of the brain [Figure 6e].
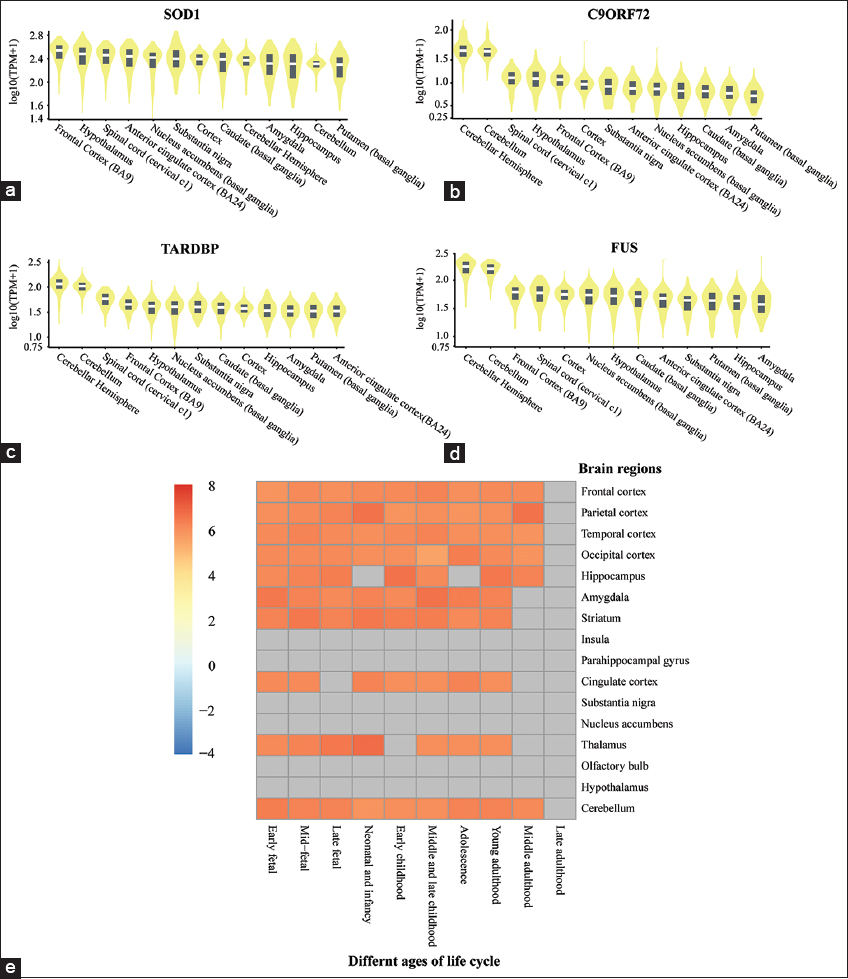 | Figure 6: Expression level of amyotrophic lateral sclerosis (ALS) high-risk genes in brain using GTEx platform. (a) Expression levels of SOD1 in different regions of the brain. (b) Expression levels of Chromosome 9 open reading frame 72 in different regions of the brain. (c) Expression levels of TAR DNA-binding protein 43 in different regions of the brain. (d) Expression levels of FUS in different regions of the brain and (e) Spatio-temporal expression heatmap from best tool showed the expression of ALS high-risk genes in brain with different age groups. [Click here to view] |
4. DISCUSSION
With the advancement of experimental technology, various computational tools and databases are available that are associated with enormous information allowing us to identify potential genes and protein, other biomolecules associated with ALS thus providing an insight to the pathogenesis. Understanding how ALS-hr-Gs regulated is therefore important in developing treatments. One of the significant approaches to find the reason for this is to identify the transcriptional and post-transcriptional regulators which are interconnected to each other and regulates functions. This is how a combinatorial regulatory network is thought to determine the human transcriptome. Here, our study focused on four ALS-hr-Gs which can be targeted by some candidate miRNAs and TFs during the process of transcription and post-transcriptional level respectively. Understanding the regulatory network and its topological features contributes in identifying the functional role of target-associated TFs and miRNAs, which may provide novel therapeutic targets, e.g., using gene expression data, a gene-regulated analysis identified potential candidate genes for squamous lung cancer [53]. Several bioinformatic integrative analyses identified key target genes, miRNA, and TF as prostate cancer signatures [54], neurodegenerative diseases [17,55]. In our study, nine miRNAs (miR-1468-3p, miR-3163, miR-33a-3p, miR-3691-3p, miR-4422, miR-4666a-3p, miR-513a-5p, miR-6504-3p, and miR-6835-3p) were commonly targeted all four ALS-hr-Gs. The coherent and in-coherent FFLs analysis showed that SOD1 and FUS directly or indirectly regulated by TFs (SP1, EGR1, and CEBPD) and TBP respectively. These coherent and in-coherent coherent structure can play an important role in the dynamic behavior of biological networks [56,57]. The structure of several coherent FFLs can be viewed as a type of redundancy engineering for biological robustness, similar to how alternate routine pathways aid competition and survival in changing environments [58]. MicroRNAs, one of the most important endogenous epigenetic biomolecules, limit target gene post-transcriptional expression [59]. A single miRNA can interact with several genes. miRNA expression is highly tissue- and cell-specific and can be used as disease diagnosis and treatment [60]. miRNAs can regulate more than 50% of coding genes and help to stable the biological processes. As a result, it is suspected that miRNAs may have a role in neurodegenerative disorders [61,62]. In this study, two miRNAs (mir-3163 and miR-4422) important for the combinatorial ALS-hr-Gs network and can be the potential target for therapeutic intervention of ALS disease. The one miRNA (mir-3163) targeted other 26 ALS-genes and also presence at 3-node motif level and can be used to target multiples genes to suppress the overlapped pathogenesis of ALS. The GO functions (BP, MF, and CC), pathways and disease association stated the role of candidate miRNAs in neuronal activity and pathways in cancers. miR-33a-3p and miR-422 were highly expressed in brain as compared to the other candidates. The expression data of ALS-hr-Gs showed continuous expression in most of the areas of brain, throughout the life cycle mostly in frontal cortex and cerebellum part of the brain. It was reported that miRNA played an important role in several molecular pathways contribute to ALS pathogenesis such as neurodegeneration and apoptosis [52]. Utilization of miRNAs/TFs and their association with genes could be a biomarker for diagnosis and clinical care of ALS patients is still in its early stages required intensive research.
5. CONCLUSION
In this study, to unravel the signaling challenges associated with ALS and regulatory mechanisms. A single protein or other biomolecule may rarely act alone to perform a specific function among living organisms. Instead, a BP inside a cell is the result of a complex series of interactions between multiple biomolecules. The structure and topology of MINs can be used to identify biomolecules involved with biological processes. Here, constructed a combinatorial ALS-hr-Gs network consisted of transcription and post-transcriptional regulators (miRNAs) and explore the regulatory mechanism of ALS-hr-Gs up and regulated by TFs and miRNAs [Table 6]. It creates a scaffold combinatorial gene regulatory network that enables systematic research on the regulation of ALS genes. The connection among ALS-hr-Gs-miRNA-TFs could be a critical three-node motif of the network. Our impended pipeline can be expanded to discover conditional combinatorial regulatory landscapes correlating to distinct cellular situations. The identified potential regulators may be experimentally validated and used as a therapeutic biomarker for ALS-disease diagnosis.
Table 6: Highlighting the amyotrophic lateral sclerosis-hr-Gs regulated by transcription factors and miRNAs.
| Name of ALS-hr-Gs | Type of regulation TF→ALS hr-Gs | Common miRNAs involved in inhibition |
|---|---|---|
| SOD1 | EGR1* SP1*# CEBPD* | miR-3163, miR-4422 |
| FUS | TBP# | miR-3163, miR-4422 |
miRNAs: microRNAs, ALS: Amyotrophic lateral sclerosis, SOD1: Superoxide dismutase 1, FUS: Fused in sarcoma, ALS-hr-Gs: Amyotrophic lateral sclerosis high-risk genes,
*: Up-regulate, #: Down-regulate, *#: Up and down-regulate
6. ACKNOWLEDGMENT
R.K. gratefully acknowledges the council for scientific and industrial research for providing research fellowship. S.H. acknowledges life science research board, defense research and development organization metcalfe house Delhi-1100054, Ministry of defence of the government of India, for support. Sincerely thank the Jaypee institute of information technology, Noida, Sec-62, Uttar Pradesh, India.
7. AUTHORS’ CONTRIBUTIONS
Rupesh Kumar; Concept and design, Data curation and acquisition, Data analysis/interpretation, Drafting manuscript, Statistical analysis, Critical revision of manuscript, Final approval. Pammi Gauba; Supervision, Critical revision of manuscript, Final approval. Shazia Haider; Concept and design and Supervision, Critical revision of manuscript, Final approval.
8. FUNDING
No funding received
9. CONFLICTS OF INTEREST
The authors report no financial or any other conflicts of interest in this work.
10. ETHICAL APPROVALS
This study does not involve experiments on animals or human subjects.
11. DATA AVAILABILITY
Data in the form of Supplementary Tables is available at publisher’s website.
12. Publisher’s Note
This journal remains neutral with regard to jurisdictional claims in published institutional affiliation.
REFERENCES
1. Rowland LP, Shneider NA. Amyotrophic lateral sclerosis. N Engl J Med2001;344:1688-700 [https://doi.org/10.1056/NEJM200105313442207]
2. Akbari Dilmaghani N, Hussen BM, Nateghinia S, Taheri M, Ghafouri-Fard S. Emerging role of microRNAs in the pathogenesis of amyotrophic lateral sclerosis. Metab Brain Dis 2021;36:737-49. [https://doi.org/10.1007/s11011-021-00697-5]
3. Bonafede R, Mariotti R. ALS pathogenesis and therapeutic approaches:The role of mesenchymal stem cells and extracellular vesicles. Front Cell Neurosci2017;11:80. [https://doi.org/10.3389/fncel.2017.00080]
4. Smukowski SN, Maioli H, Latimer CS, Bird TD, Jayadev S, Valdmanis PN. Progress in amyotrophic lateral sclerosis gene discovery:Reflecting on classic approaches and leveraging emerging technologies. Neurol Genet2022;8:e669. [https://doi.org/10.1212/NXG.0000000000000669]
5. Boylan K. Familial amyotrophic lateral sclerosis. Neurol Clin2015;33:807-30. [https://doi.org/10.1016/j.ncl.2015.07.001]
6. Martin S, Al Khleifat A, Al-Chalabi A. What causes amyotrophic lateral sclerosis?F1000Res2017;6:371. [https://doi.org/10.12688/f1000research.10476.1]
7. Mathis S, Goizet C, Soulages A, Vallat JM, Masson GL. Genetics of amyotrophic lateral sclerosis:A review. J Neurol Sci 2019;399:217-26. [https://doi.org/10.1016/j.jns.2019.02.030]
8. Volk AE, Weishaupt JH, Andersen PM, Ludolph AC, Kubisch C. Current knowledge and recent insights into the genetic basis of amyotrophic lateral sclerosis. Med Genet 2018;30:252-8. [https://doi.org/10.1007/s11825-018-0185-3]
9. Joilin G, Leigh PN, Newbury SF, Hafezparast M. An overview of MicroRNAs as biomarkers of ALS. Front Neurol2019;10:186. [https://doi.org/10.3389/fneur.2019.00186]
10. Goutman SA, Chen KS, Paez-Colasante X, Feldman EL. Emerging understanding of the genotype-phenotype relationship in amyotrophic lateral sclerosis. Handb Clin Neurol 2018;148:603-23. [https://doi.org/10.1016/B978-0-444-64076-5.00039-9]
11. Kumar R, Haider S. The current genetics of amyotrophic lateral sclerosis (ALS):Since 2015. Int J Biotechnol Pharm Sci2022;11:105-12.
12. Kumar R, Malik Z, Singh M, Mani S, Ponnusamy K, Haider S. Amyotrophic lateral sclerosis risk genes and suppressor. Curr Gene Ther2023;23:148-62. [https://doi.org/10.2174/1566523223666221108113330]
13. Abramzon YA, Fratta P, Traynor BJ, Chia R. The overlapping genetics of amyotrophic lateral sclerosis and frontotemporal dementia. Front Neurosci 2020;14:42. [https://doi.org/10.3389/fnins.2020.00042]
14. Ghasemi M, Brown RH Jr. Genetics of amyotrophic lateral sclerosis. Cold Spring Harb Perspect Med2018;8:a024125. [https://doi.org/10.1101/cshperspect.a024125]
15. Morgan S, Orrell RW. Pathogenesis of amyotrophic lateral sclerosis. Br Med Bull 2016;119:87-98. [https://doi.org/10.1093/bmb/ldw026]
16. Tu K, Yu H, Hua YJ, Li YY, Liu L, Xie L, et al. Combinatorial network of primary and secondary microRNA-driven regulatory mechanisms. Nucleic Acids Res 2009;37:5969-80. [https://doi.org/10.1093/nar/gkp638]
17. Su LN, Song XQ, Xue ZX, Zheng CQ, Yin HF, Wei HP. Network analysis of microRNAs, transcription factors, and target genes involved in axon regeneration. J Zhejiang Univ Sci B2018;19:293-304. [https://doi.org/10.1631/jzus.B1700179]
18. Huntzinger E, Izaurralde E. Gene silencing by microRNAs:Contributions of translational repression and mRNA decay. Nat Rev Genet2011;12:99-110. [https://doi.org/10.1038/nrg2936]
19. De Felice B, Guida M, Guida M, Coppola C, De Mieri G, Cotrufo R. A miRNA signature in leukocytes from sporadic amyotrophic lateral sclerosis. Gene 2012;508:35-40. [https://doi.org/10.1016/j.gene.2012.07.058]
20. Angelucci F, Cechova K, Valis M, Kuca K, Zhang B, Hort J. MicroRNAs in alzheimer's disease:Diagnostic markers or therapeutic agents?Front Pharmacol 2019;10:665. [https://doi.org/10.3389/fphar.2019.00665]
21. Chen X, Ba Y, Ma L, Cai X, Yin Y, WangK, et al. Characterization of microRNAs in serum:A novel class of biomarkers for diagnosis of cancer and other diseases. Cell Res 2008;18:997-1006. [https://doi.org/10.1038/cr.2008.282]
22. Jin XF, Wu N, Wang L, Li J. Circulating microRNAs:A novel class of potential biomarkers for diagnosing and prognosing central nervous system diseases. Cell Mol Neurobiol 2013;33:601-13. [https://doi.org/10.1007/s10571-013-9940-9]
23. Di Pietro L, Baranzini M, Berardinelli MG, Lattanzi W, Monforte M, TascaG, et al. Potential therapeutic targets for ALS:MIR206, MIR208b and MIR499 are modulated during disease progression in the skeletal muscle of patients. Sci Rep2017;7:9538. [https://doi.org/10.1038/s41598-017-10161-z]
24. Kovanda A, Leonardis L, Zidar J, Koritnik B, Dolenc-Groselj L, Kovacic SR, et al. Differential expression of microRNAs and other small RNAs in muscle tissue of patients with ALS and healthy age-matched controls. Sci Rep 2018;8:5609. [https://doi.org/10.1038/s41598-018-23139-2]
25. De Andrade HM, de Albuquerque M, Avansini SH, de S Rocha C, Dogini DB, NucciA, et al. MicroRNAs-424 and 206 are potential prognostic markers in spinal onset amyotrophic lateral sclerosis. J Neurol Sci2016;368:19-24. [https://doi.org/10.1016/j.jns.2016.06.046]
26. Sheinerman KS, Toledo JB, Tsivinsky VG, Irwin D, Grossman M, WeintraubD, et al. Circulating brain-enriched microRNAs as novel biomarkers for detection and differentiation of neurodegenerative diseases. Alzheimers Res Ther2017;9:89. [https://doi.org/10.1186/s13195-017-0316-0]
27. Toivonen JM, Manzano R, Olivan S, Zaragoza P, Garcia-Redondo A, Osta R. MicroRNA-206:A potential circulating biomarker candidate for amyotrophic lateral sclerosis. PLoS One2014;9:e89065. [https://doi.org/10.1371/journal.pone.0089065]
28. Aumiller V, Forstemann K. Roles of microRNAs beyond development--metabolism and neural plasticity. Biochim Biophys Acta 2008;1779:692-6. [https://doi.org/10.1016/j.bbagrm.2008.04.008]
29. Carissimi C, Fulci V, Macino G. MicroRNAs:Novel regulators of immunity. Autoimmun Rev 2009;8:520-4. [https://doi.org/10.1016/j.autrev.2009.01.008]
30. Quinlan S, Kenny A, Medina M, Engel T, Jimenez-Mateos EM. MicroRNAs in neurodegenerative diseases. Int Rev Cell Mol Biol2017;334:309-43. [https://doi.org/10.1016/bs.ircmb.2017.04.002]
31. Rajgor D. Macro roles for microRNAs in neurodegenerative diseases. Noncoding RNA Res 2018;3:154-9. [https://doi.org/10.1016/j.ncrna.2018.07.001]
32. Faraoni I, Antonetti FR, Cardone J, Bonmassar E. miR-155 gene:A typical multifunctional microRNA. Biochim Biophys Acta 2009;1792:497-505. [https://doi.org/10.1016/j.bbadis.2009.02.013]
33. Junker A, Krumbholz M, Eisele S, Mohan H, Augstein F, BittnerR, et al. MicroRNA profiling of multiple sclerosis lesions identifies modulators of the regulatory protein CD47. Brain2009;132:3342-52. [https://doi.org/10.1093/brain/awp300]
34. Wang WX, Visavadiya NP, Pandya JD, Nelson PT, Sullivan PG, Springer JE. Mitochondria-associated microRNAs in rat hippocampus following traumatic brain injury. Exp Neurol 2015;265:84-93. [https://doi.org/10.1016/j.expneurol.2014.12.018]
35. Agarwal V, Bell GW, Nam JW, Bartel DP. Predicting effective microRNA target sites in mammalian mRNAs. Elife 2015;4:e05005. [https://doi.org/10.7554/eLife.05005]
36. Huang HY, Lin YC, Li J, Huang KY, Shrestha S, HongHC, et al. miRTarBase 2020:Updates to the experimentally validated microRNA-target interaction database. Nucleic Acids Res2020;48:D148-54. [https://doi.org/10.1093/nar/gkz896]
37. Chen Y, Wang X. miRDB:An online database for prediction of functional microRNA targets. Nucleic Acids Res2020;48:D127-31. [https://doi.org/10.1093/nar/gkz757]
38. Han H, Shim H, Shin D, Shim JE, Ko Y, ShinJ, et al. TRRUST:A reference database of human transcriptional regulatory interactions. Sci Rep2015;5:11432. [https://doi.org/10.1038/srep11432]
39. Tong Z, Cui Q, Wang J, Zhou Y. TransmiR v2.0:An updated transcription factor-microRNA regulation database.Nucleic Acids Res 2019;47:D253-8. [https://doi.org/10.1093/nar/gky1023]
40. Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, et al. Cytoscape:A software environment for integrated models of biomolecular interaction networks. Genome Research 2003;13:2498-50. [https://doi.org/10.1101/gr.1239303]
41. El Baroudi M, Cora D, Bosia C, Osella M, Caselle M. A curated database of miRNA mediated feed-forward loops involving MYC as master regulator. PLoS One 2011;6:e14742. [https://doi.org/10.1371/journal.pone.0014742]
42. Huang JC, Babak T, Corson TW, Chua G, Khan S, GallieBL, et al. Using expression profiling data to identify human microRNA targets. Nat Methods2007;4:1045-9. [https://doi.org/10.1038/nmeth1130]
43. Godard P, van Eyll J. Pathway analysis from lists of microRNAs:Common pitfalls and alternative strategy. Nucleic Acids Res2015;43:3490-7. [https://doi.org/10.1093/nar/gkv249]
44. Liao Y, Wang J, Jaehnig EJ, Shi Z, Zhang B. WebGestalt 2019:Gene set analysis toolkit with revamped UIs and APIs. Nucleic Acids Res 2019;47:W199-205. [https://doi.org/10.1093/nar/gkz401]
45. Kavakiotis I, Alexiou A, Tastsoglou S, Vlachos IS, Hatzigeorgiou AG. DIANA-miTED:A microRNA tissue expression database. Nucleic Acids Res 2022;50:D1055-61. [https://doi.org/10.1093/nar/gkab733]
46. GTEx Consortium. Human genomics. The genotype-tissue expression (GTEx) pilot analysis:Multitissue gene regulation in humans. Science 2015;348:648-60. [https://doi.org/10.1126/science.1262110]
47. Guo L, Lin W, Zhang Y, Li W, Wang J. BEST:A web server for brain expression Spatio-temporal pattern analysis. BMC Bioinformatics 2019;20:632. [https://doi.org/10.1186/s12859-019-3222-6]
48. Garzon R, Marcucci G, Croce CM. Targeting microRNAs in cancer:Rationale, strategies and challenges. Nat Rev Drug Discov2010;9:775-89. [https://doi.org/10.1038/nrd3179]
49. Inui M, Martello G, Piccolo S. MicroRNA control of signal transduction. Nat Rev Mol Cell Biol 2010;11:252-63. [https://doi.org/10.1038/nrm2868]
50. Ramirez-Moya J, Santisteban P. miRNA-directed regulation of the main signaling pathways in thyroid cancer. Front Endocrinol (Lausanne) 2019;10:430. [https://doi.org/10.3389/fendo.2019.00430]
51. Kim H, Iizasa H, Kanehiro Y, Fekadu S, Yoshiyama H. Herpesviral microRNAs in cellular metabolism and immune responses. Front Microbiol2017;8:1318. [https://doi.org/10.3389/fmicb.2017.01318]
52. Piedade D, Azevedo-Pereira JM. The role of microRNAs in the pathogenesis of herpesvirus infection. Viruses 2016;8:156. [https://doi.org/10.3390/v8060156]
53. Bai J, Hu S. Transcriptome network analysis reveals potential candidate genes for squamous lung cancer. Int J Mol Med 2012;29:95-101. [https://doi.org/10.3892/ijmm.2011.796]
54. Mangangcha IR, Malik MZ, Kucuk O, Ali S, Singh RK. Kinless hubs are potential target genes in prostate cancer network. Genomics2020;112:5227-39. [https://doi.org/10.1016/j.ygeno.2020.09.033]
55. Karnati HK, Panigrahi MK, Gutti RK, Greig NH, Tamargo IA. miRNAs:Key players in neurodegenerative disorders and epilepsy. J Alzheimers Dis 2015;48:563-80. [https://doi.org/10.3233/JAD-150395]
56. Kalir S, Mangan S, Alon U. A coherent feed-forward loop with a SUM input function prolongs flagella expression in Escherichia coli. Mol Syst Biol2005;1:2005.0006. [https://doi.org/10.1038/msb4100010]
57. Mangan S, Zaslaver A, Alon U. The coherent feedforward loop serves as a sign-sensitive delay element in transcription networks. J Mol Biol 2003;334:197-204. [https://doi.org/10.1016/j.jmb.2003.09.049]
58. Le DH, Kwon YK. A coherent feedforward loop design principle to sustain robustness of biological networks. Bioinformatics2013;29:630-7. [https://doi.org/10.1093/bioinformatics/btt026]
59. O'Brien J, Hayder H, Zayed Y, Peng C. Overview of MicroRNA biogenesis, mechanisms of actions, and circulation. Front Endocrinol (Lausanne)2018;9:402. [https://doi.org/10.3389/fendo.2018.00402]
60. Condrat CE, Thompson DC, Barbu MG, Bugnar OL, Boboc A, CretoiuD, et al. miRNAs as biomarkers in disease:Latest findings regarding their role in diagnosis and prognosis. Cells2020;9:276. [https://doi.org/10.3390/cells9020276]
61. Gascon E, Gao FB. Cause or effect:Misregulation of microRNA pathways in neurodegeneration. Front Neurosci2012;6:48. [https://doi.org/10.3389/fnins.2012.00048]
62. Krol J, Loedige I, Filipowicz W. The widespread regulation of microRNA biogenesis, function and decay. Nat Rev Genet2010;11:597-610. [https://doi.org/10.1038/nrg2843]