
1. INTRODUCTION
Nowadays, applying convenient and inexpensive read-out technologies like the nucleic acid lateral flow immunoassay (NALFIA), could simplify the process rather in the reduced time and avoidance of hazardous chemicals [1]. It can avoid using gel electrophoresis (GE). NALFIA using the lateral flow dipstick (LFD) combined with polymerase chain reaction (PCR) employing labeled specific primers was introduced for detecting specific amplified DNA target, so called PCR-NALFIA. PCR-NALFIA is a quick and simple diagnostic technique that is easily incorporated into clinical diagnostics. When compared to conventional detection techniques, the lateral flow strips have advantages in terms of speed and simplicity [2]. Using an antibody capture line and a labeled amplicon, nucleic acids are trapped on the LFD and transformed into an antibody-dependent format. Mechanically, PCR can be used to amplify the desired DNA target by utilizing specific primers labeled biotin and carboxyfluorescein (FAM). The analyte that is contained the biotin-FAM labeled amplicon is identified by binding to an anti-fluorescein antibody previously sprayed on a nitrocellulose membrane and AuNPs labeled with anti-FAM antibody are used as reporter that enables the colored visualization. According to the analyte concentration, the response is directly proportional [3]. PCR-NALFIA assay is a viable tool for species-specific detection with good accuracy [4,5]. NALFIA has already been utilized as a biosensor for identifying specific amplified target genes [6] and has been broadly applied for point of care (POC) diagnostic tools. At present, this assay has also crossed other research areas [1–3]. PCR-NALFIA may be an alternatively valuable technique for manipulating DNA evidence from biological evidence during a criminal investigation. This technique can easily and quickly facilitate determination of human gender which is crucial in forensic casework such as murder, rape, and missing persons at the crime scene [7]. Sex identification based on DNA analysis is important because forensic and archaeological samples devoid of morphological diagnostic characteristics are ineligible for morphological sex determination [8]. Human sex determination usually relies on two genes that are sex-determining region Y (SRY) and amelogenin (AMEL) on nuclear DNA [9-11]. Several PCR-based methods, SRY gene has been used as a genetic marker [12,13]. Several methods based on detection of the AMEL gene have been reported in forensic science [8,14]. The loop-mediated isothermal amplification (LAMP) assay has been reported for human sex identification using AMEL locus [15]. To avoid GE, the LAMP-LFD has been explored for detecting human male DNA based on SRY gene [16]. In addition, the SRY gene, as an alternative Y-specific marker, is an adjunct with the AMEL gene. The incorrect identification of types of some males as females due to the frequent occurrence of large area Y chromosomal deletions, which lead to AMELY allelic dropout and inaccurate inferences about the sex of the DNA source [14,17]. Reliable and rapid detection of DNA among molecular analyses is increasingly significant. The advancement of molecular biology and nanotechnology has attracted a great deal of interest in recent years in the field of bioanalytical sciences, and it has been reviewed as having a considerable impact on the ability to accurately and precisely detect a range of target analytes without the use of specialized instrumentation [18,19]. In the medical field, there should only rarely be any diagnostic doubt left following a full clinical and radiologic assessment. When necessary, molecular testing is basic. For example, achondroplasia (ACH), which is the most common inherited skeletal dysplasia caused by the mutation in the fibroblast growth factor receptor 3 (FGFR3) gene. The ACH can be identified by its radiological and clinical characteristics. However, molecular testing for FGFR3 gene mutations can still be helpful for patients who cannot be separated from hypochondroplasia (HCH). In addition, it is essential for prenatal diagnostic confirmation. Around 98% of persons with ACH contain a c.1138G > A gene change, and 1% or so carry a c.1138G > C mutation [20]. Two mutations in the FGFR3 gene at nucleotide c.1138 result in a p.Gly380Arg substitution that is responsible for most cases of ACH [21]. However, a significant number of HCH cases has no mutations in FGFR3, indicating that other genes may be involved in this phenotype [22]. To distinguish ACH from other types of severe dwarfism, particularly HCH, when prenatal dwarfism is suspected, the detection of the FGFR3 gene mutations is crucial. These mutations can be identified by a variety of molecular methods. There are several methods for detecting a G1138A mutation in the FGFR3 gene such as DNA sequencing [23], restriction fragment length polymorphism-PCR (RFLP-PCR) [24], denatured high-performance liquid chromatography (dHPLC) [25], real-time PCR [26], high-resolution melting (HRM) analysis [27], and allele-specific PCR (AS-PCR) [28], Sanger sequencing was employed to confirm these mutations; however, HRM is a quicker, more accurate, and less expensive genotyping assay [29]. Many approaches for identifying target genes have been developed. Nevertheless, conventional screenings are unable to always detect target genes that are present in uncommon conditions. Several DNA-based assays for gene identification have been designed in order to address some of the shortcomings of the conventional approaches. Despite providing strong specificity and sensitivity, they are not frequently used in molecular diagnosis programs due to complicated procedures or lack of funding. The PCR-NALFIA system offers a quick and easy detection technology for target gene verification. This detection format can be read within 5 min on the LFD strip by the naked eye. To the best of our knowledge, the PCR-NALFIA assay has not yet been used to detect human DNA and determine human gender based on human SRY gene. Moreover, the AS-PCR-NALFIA assay also has not yet been used to detect G1138A mutation in the FGFR3 gene for diagnosis of ACH. Therefore, the objective of the present study aimed to detect human DNA and gender from various biological evidence types such as blood, semen, and saliva samples as well as to detect the G1138A mutation in the FGFR3 gene from DNA samples for diagnosis of ACH that is more rapid and convenient technique than the previous methods.
2. MATERIALS AND METHODS
2.1. DNA Template Preparation
The DNA templates used in this research were provided into two parts for detection of the SRY gene and the G1138A mutation in the FGFR3 gene. The DNA templates for detection of the SRY gene were prepared from blood, semen and saliva samples. Male blood samples (n = 5) were kindly obtained from Blood Bank Srinagarind hospital, Khon Kaen University, Khon Kaen, Thailand and stored at 4°C. Semen samples (n = 10) were received from Infertility clinic, Srinagarind hospital and temporarily stored at 37°C. Male saliva samples (n = 5) were collected from unrelated volunteers and stored at 37°C. Moreover, female blood (n = 5) and female saliva (n = 5) samples were also provided and used as negative controls for specificity analysis because SRY gene cannot be found in female. The genomic DNA was extracted by using GF-1 Blood DNA Extraction Kit (Vivantis, Malaysia) for blood samples and Illustra tissue & cells genomicPrep Mini Spin Kit (GE Healthcare, USA) for semen and saliva samples following to the manufacturer’s protocols. For the DNA templates for detection of the G1138A mutation in the FGFR3 gene, the DNA samples (n = 5) used in this research were kindly supplied from genetics and molecular medicine laboratory, Department of Biochemistry, Faculty of Medicine, Khon Kaen university, Khon Kaen, Thailand. Briefly, genomic DNA extracted from peripheral blood of patients with a clinical diagnosis of ACH using isopropanol-fractionation with concentrated NaI and SDS technique. These DNA solutions were used as a template for PCR and allele-specific PCR-NALFIA amplification. The Khon Kaen University Ethics Committee for Human Research had granted ethical approval for the research (HE631576).
2.2. Primer Design and Preparation
The SRY primers used in this research were designed using the Primer-BLAST software (https://www.ncbi.nlm.nih.gov/tools/primer-blast/index.cgi) based on the human SRY gene (accession number: JQ811934.1). The specificity of the primers was assessed by using the BLAST software (https://blast.ncbi.nlm.nih.gov/Blast.cgi) against the NCBI database to ensure that no homology exists with other sequences. The ACH primers used in this research were applied from the previous report [28]. The specific forward and reverse primers of both genes were modified by 5′ labeling with biotin and FAM respectively. Primers for PCR are listed in Table 1. All primers were synthesized by Macrogen (Soul, Korea).
Table 1: The primers used in this study.
| Gene | Primer | 5′-Sequence-3′ | Product size (bp) | References |
|---|---|---|---|---|
| SRY | SRYF-BIOTIN | BIOTIN-AACAGTAAAGGCAACGTCCA | 220 | Present study |
| SRYR-FAM | FAM-ATTTCTCTCTGTGCATGGCC | |||
| SRYF | AACAGTAAAGGCAACGTCCA | |||
| SRYR | ATTTCTCTCTGTGCATGGCC | |||
| FGFR3 | ACHF-BIOTIN | BIOTIN-GCATCCTCAGCTACA | 122 | [28] |
| ACHR-FAM | FAM-GGAGATCTTGTGCACGGTGG | |||
| ACHF | GCATCCTCAGCTACA | |||
| ACHR | GGAGATCTTGTGCACGGTGG |
2.3. PCR Amplification for PCR-NALFIA
To get the SRY and FGFR3 genes, the labeled specific primers of both genes were performed in the PCR-NALFIA assay. For the SRYF-BIOTIN and SRYR-FAM primers producing fragments of 220 bp, the PCR mixture was carried out in a 25 μL reaction mixture composed of 1× PCR supermix (OnePCR Ultra, Bio-Helix), 0.2 μM of each primer, 600 mM betaine (Sigma-Aldrich, USA), as well as 100 ng of each DNA template and sterilized deionized water to make up a final volume of 25 μL. The thermal cycling was performed using thermal cycler (Prima 96™, Himedia) with the conditions consisted of pre-heating at 94 °C for 5 min, 25 cycles of consecutive incubations at 94 °C for 30 sec, 63 °C for 30 sec, and 72 °C for 30 sec, followed by a final extension at 72 °C for 5 min. For the ACHF-BIOTIN and ACHR-FAM primers producing fragments of 122 bp, the ASPCR reaction was also the same as described above and was cycled under the conditions previously described above. All PCR products were purified by using a PCR purification kit (Invitrogen by Thermo Fisher Scientific, USA).
2.4. PCR-NALFIA
Following PCR amplification, the resultant double labeled PCR product was separated into 2 parts for GE and NALFIA system analysis. For GE system, 10 μL of the double labeled PCR products were examined on 1.5% agarose gel using 0.5× TBE buffer running in voltage 100 V for 30 min stained with fluorescent dye (Smobio, Taiwan) and visualized under the UVITECH gel documentation system. The double labeled PCR products were compared with 100 bp DNA Ladder RTU (GeneDireX, USA). For NALFIA system, 10 μL of the double labeled PCR products were mixed with 100 μL of the HybriDetect assay buffer. Then, the universal LFD (Milenia Genline HybriDetect 1, Giessen, Germany) was dipped into the mixture for 5 min to detect the specific double labeled PCR product. Within 5 min, the results were interpreted by the naked eye on the LFD. Test band and control band are clearly formed the color bands. This indicated that the double labeled PCR amplicon was detected (positive). In contrast, only the control band is visible as the color band. It indicated no double labeled PCR amplicon was detected (negative). In addition, PCR-NALFIA dipsticks were photographed by digital camera (Nikon D5100 with AF-S Micro NIKKOR 105mm f/2.8).
2.5. Sensitivity of PCR-NALFIA Assay
To determine the sensitivity of PCR- and AS-PCR-NALFIA assays for both SRY and FGFR3 genes, different concentrations of the DNA template (100, 50, 25, 10, 5, 1, 0.1, or 0.01 ng/reaction) were prepared and used in PCR and then simultaneously evaluated by the LFD. In both genes, each experiment was repeated three times.
2.6. Specificity of PCR-NALFIA Assay
For SRY gene detection, since the SRY gene is presented only on male DNA, the specificity of the designed primers in the PCR-NALFIA reaction for SRY gene detection was performed to confirm positive and negative results with male and female DNA samples, respectively. Twenty-five nanograms of male or female genomic DNA were used in PCR-NALFIA reactions. For consistency of the experiment, male DNA was extracted from blood, semen and saliva while a sample of the female DNA was extracted from both blood and saliva. Sex determination was also examined. For the detection of G1138A mutation in the FGFR3 gene for diagnosis of ACH, the specificity of labeled primers was performed to confirm positive and negative results with ACH patient and normal individual DNA samples, respectively. Twenty-five nanograms of DNA sample were used in AS-PCR-NALFIA reactions. Moreover, the PCR products in the present study were analyzed to confirm the PCR product size and sequence by using DNA sequencing (Macrogen Inc., Korea).
3. RESULTS
3.1. PCR-NALFIA
A schematic overview of the conceptual basis of the PCR-NALFIA assay for human SRY gene detection is displayed in Figure 1. In PCR-NALFIA assay, after the PCR reaction, if the specific double labeled PCR product is present forming a biotin/FAM labeled amplicon. The double-labeled amplicon is mixed with the HybriDetect assay buffer, and dipped the sample pad of the LFD into the assay buffer. Via capillarity, the amplicon moves toward the direction of the wicking pad. FAM-specific antibodies attached to the membrane of the LFD recognize the FAM molecule. A biotin-specific ligand on the test line of the LFD detects the biotin on the opposite side of the amplicon and blocks it, producing a red-blue band. A control line is colored when an excess of gold nanoparticles is blocked over it by an antibody that is recognized by particular anti-rabbit antibodies. If the test and control bands form the color bands, it indicates that the double-labeled PCR amplicon was detected (positive). In contrast, if only the control band forms visible color bands, it indicates that no PCR amplicon was detected (negative). For SRY gene detection, male and female DNA samples were used as a template to amplify the human SRY gene. The SRY gene is located only on the Y chromosome in males, and there is no SRY gene in females. The results showed that the double-labeled PCR products were detected by NALFIA assay but not GE. The universal LFD showed the color at both the test and control lines in the male DNA sample. In contrast, the LFD from female DNA only displayed negative results, showing the color only at the control line. The reactions also included a negative control with no DNA template, whose output was negative, as shown in Figure 2a. On GE analysis, the specific double-labeled amplicon was invisible. However, a positive response was achieved by employing the specific double-labeled amplicon in the NALFIA system as an analyte. In order to verify the existence of the targeted double-labeled amplicon in the PCR-NALFIA reaction, regular primers were used to amplify the human SRY gene utilizing the double-labeled amplicon as a template. As expected, the 220 bp products of SRY fragments were detectable when using the double-labeled amplicon as a template in the PCR reaction. These results indicated that double-labeled amplicons are presented in the reaction for LFD testing. However, PCR products generated from normal primers only showed the band at the control line on the LFD, although 220 bp fragments of SRY gene are presented with male DNA. Moreover, DI (deionized) water was tested in the LFD, and its response was also negative, as shown in Figure 2b. The conceptual basis of the AS-PCR-NALFIA assay for detecting the heterozygous G1138A mutation in the FGFR3 gene is presented in Figure 3. In AS-PCR-NALFIA assay, after ASPCR amplification, if a specific double-labeled ASPCR product is present, a biotin/FAM-labeled amplicon will be formed. If the color bands form on the test and control bands, it indicates that the double-labeled ASPCR product was detected (positive). In contrast, if only the color band is visible on the control band, it means that no ASPCR amplicon was detected (negative). Then, the new AS-PCR-NALFIA protocol for detecting the G1138A mutation of FGFR3 gene was developed as follows. The FGFR3 gene was amplified by using the set of specific primers were performed for AS-PCR-NALFIA. The DNA samples from patients with ACH (heterozygous for the G1138A mutation) and normal individuals were used as a template to amplify FGFR3 gene. After ASPCR amplification, the double-labeled PCR product was separated into two parts for GE and NALFIA system analysis. The results showed that the double-labeled ASPCR products were detected only using NALFIA but not by using GE. The LFD showed the color at both the test and control lines in individual heterozygous for the G1138A mutation. In contrast, the LFD from normal individuals was always negative, showing the color only at the control line. The reactions also included a negative control with no DNA template, and its output was negative, as shown in Figure 4a. The double labeled ASPCR product was invisible in the GE assay. However, a positive response was accomplished using the double-labeled ASPCR product in the NALFIA system as an analyte. Thus, to ensure the presence of the double-labeled ASPCR product in the reaction from AS-PCR-NALFIA with labeled primers. Therefore, the FGFR3 gene was amplified with normal primer using the double-labeled ASPCR product as a template. As expected, the 122 bp products of FGFR3 fragments were detected when using the double-labeled ASPCR product as the template in the ASPCR reaction. These results indicated that the double-labeled ASPCR products are present in the reaction for LFD testing. However, ASPCR products generated from normal primers showed the band only at the control line on the LFD, although 122 bp fragments of the FGFR3 gene are present with individual heterozygous for the G1138A mutation. Moreover, DI (deionized) water tested in the LFD also demonstrated a negative response, as shown in Figure 4b.
 | Figure 1: The conceptual basis of the polymerase chain reaction-nucleic acid lateral flow immunoassay assay for human SRY gene detection. Modified from [31]. [Click here to view] |
 | Figure 2: The polymerase chain reaction-nucleic acid lateral flow immunoassay (PCR-NALFIA) assay for detection of the human SRY gene. (a) PCR using primers SRYF/SRYR labeled with biotin and FAM, respectively, for human SRY gene detection. (b) PCR using unlabeled primers SRYF/SRYR for the detection of human SRY gene. Upper part: Agarose GE, Lower part: PCR-NALFIA assay, CL: Control line, TL: Test line. (M: 100 bp DNA marker and -ve: Negative control). [Click here to view] |
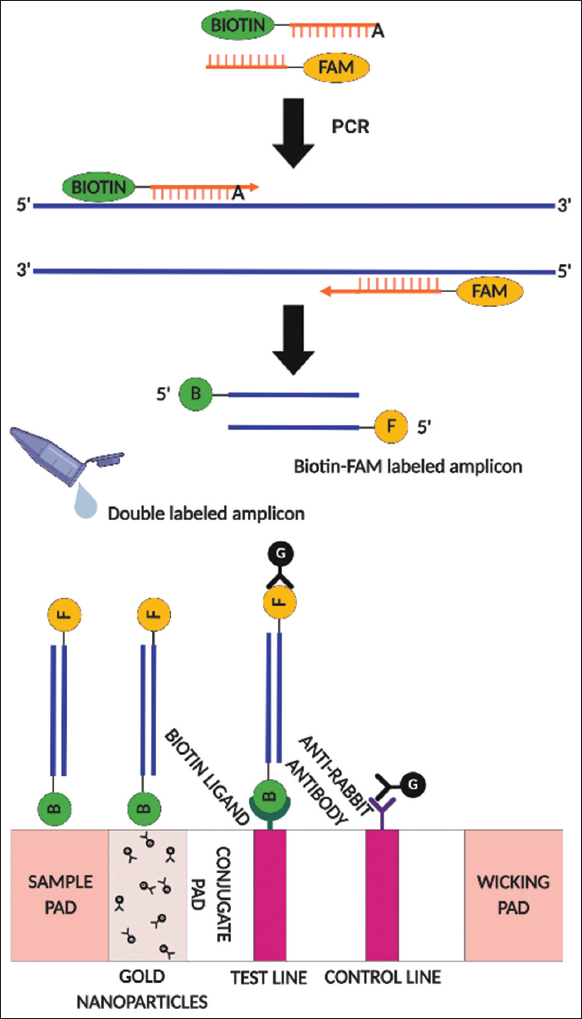 | Figure 3: The conceptual basis of the AS-polymerase chain reaction-nucleic acid lateral flow immunoassay for detection of the heterozygous G1138A mutation in the FGFR3 gene. Modified from [31]. [Click here to view] |
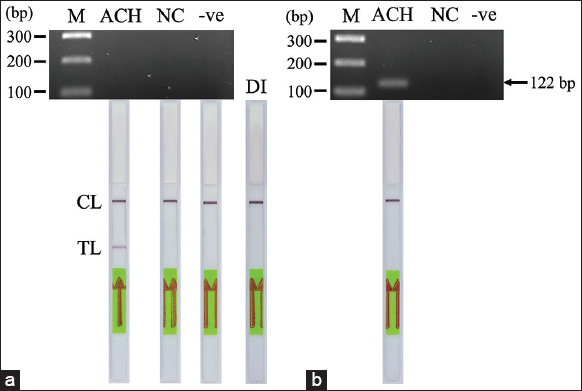 | Figure 4: The AS-polymerase chain reaction-nucleic acid lateral flow immunoassay (PCR-NALFIA) assay for detection of the heterozygous G1138A mutation in the FGFR3 gene. (a) PCR using primers ACHF/ACHR labeled with biotin and FAM, respectively, for FGFR3 gene detection. (b) PCR using unlabeled primers ACHF/ACHR for the detection of the FGFR3 gene. Upper part: Agarose GE, Lower part: AS-PCR-NALFIA assay, CL: Control line, TL: Test line. (M: 100 bp DNA marker, ACH: Achondroplasia (heterozygous for the G1138A mutation), NC: Normal individual; -ve: Negative control). [Click here to view] |
3.2. Sensitivity of NALFIA System
For SRY gene detection, to determine the lowest detection limit of this PCR-NALFIA system, PCR was performed using different concentrations of the DNA template ranging from 0.01 to 100 ng. The results showed that the human SRY gene amplified up to 1 ng of DNA template in PCR-NALFIA assay. Visual detection of a faint test line on DNA templates as low as 1 ng was seen with the LFD, as shown in Figure 5. However, as described above, the double-labeled amplicon was invisible on the agarose gel. Thus, there is no data shown. Meanwhile, this study’s detection limit of PCR-NALFIA system was 1 ng/reaction. For the detection of G1138A mutation in FGFR3 gene, to identify the detection limit of this AS-PCR-NALFIA system, ASPCR was performed using different concentrations of the DNA template ranging from 0.01 to 100 ng. The results showed that the FGFR3 gene amplified 1 ng of DNA template in AS-PCR-NALFIA assay. With the LFD, a faint test line was visually detected on DNA templates as low as 1 ng in concentration, as shown in Figure 6. ASPCR-based GE was also performed simultaneously, but the double-labeled ASPCR product was invisible on the agarose gel, as described above. Therefore, the data are not shown here. Thus, the detection limit of AS-PCR-NALFIA system in this study was 1 ng/reaction.
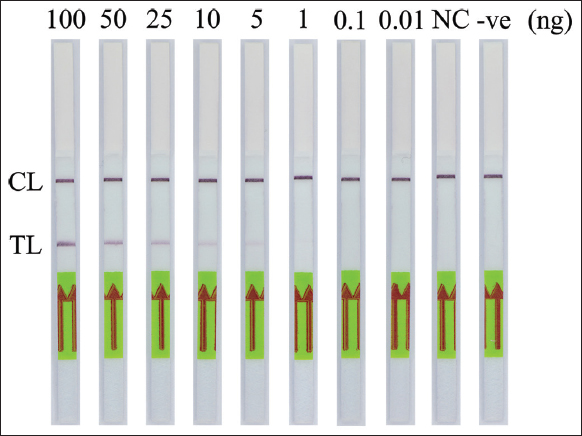 | Figure 5: The sensitivity of polymerase chain reaction-nucleic acid lateral flow immunoassay assay for detection of the human SRY gene. CL: Control line, TL: Test line (NC: Female DNA template; -ve: Negative control). [Click here to view] |
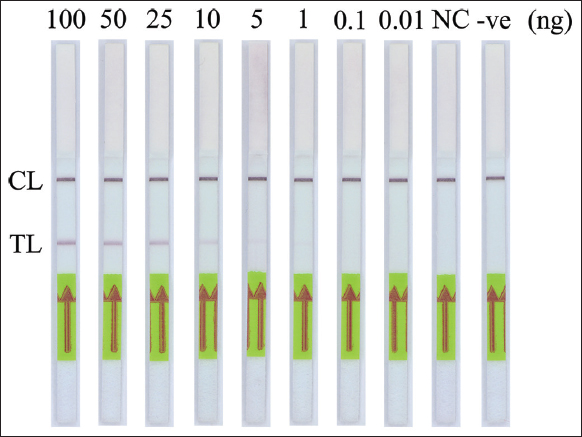 | Figure 6: The sensitivity of AS-polymerase chain reaction-nucleic acid lateral flow immunoassay assay for detection of the heterozygous G1138A mutation in the FGFR3 gene. CL: Control line, TL: Test line (NC: Normal individual; -ve: Negative control). [Click here to view] |
3.3. Specificity of NALFIA System
The specificity of the PCR-NALFIA assay for SRY gene detection was performed under the optimal PCR conditions with the genomic DNA concentration of 25 ng obtained from both males and females. After the PCR amplification was completed, the LFD was visually inspected. The applicability of DNA target amplification was confirmed by the appearance of the test band on the LFD, which was observed by the naked eye. The double-labeled amplicon of the SRY gene was detectable using NALFIA when using a male DNA as the template in the PCR-NALFIA assay. In contrast, no positive result was observed when using a female DNA as a template. The negative result is also present in the negative control (no DNA template), as shown in Figures 7-9, respectively. The result confirmed the specificity of the PCR-NALFIA assay for SRY detection. To verify the specificity of the AS-PCR-NALFIA assay, ASPCR was carried out using the genomic DNA concentration of 25 ng obtained from patients with ACH, and an ordinary individual’s DNA was used as a template to amplify FGFR3 gene. The ASPCR products, obtained by employing the methods described above, were then introduced to the NALFIA system, and the LFD was visually examined and observed by the naked eye. The results showed that the LFD revealed the color at both the test and control lines in positive control for heterozygous G1138A mutation and patients with ACH (heterozygous for the G1138A mutation). On the other hand, the LFD from an ordinary individual always demonstrates negative results, showing the color only at the control line. In addition, no DNA template was used as the negative control in the experiments, and its outcome was also negative, as shown in Figure 10.
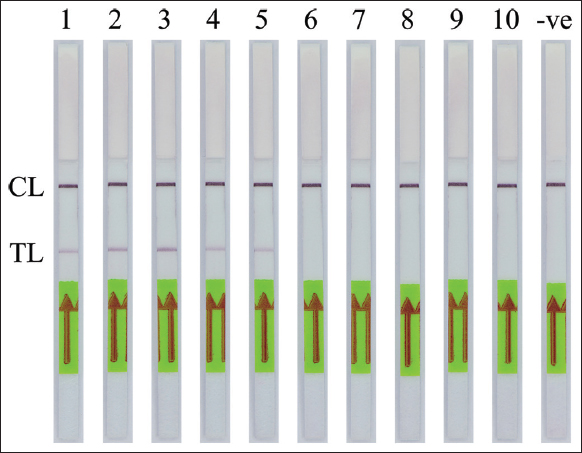 | Figure 7: The specificity of polymerase chain reaction-nucleic acid lateral flow immunoassay assay for detection of the human SRY gene from blood samples. CL: Control line, TL: Test line. (1–5: Male blood samples, 6–10: Female blood samples; -ve: Negative control). [Click here to view] |
 | Figure 8: The specificity of polymerase chain reaction-nucleic acid lateral flow immunoassay assay for detection of the human SRY gene from semen samples. CL: Control line, TL: Test line. (1–10: Semen samples, NC: Female DNA template; -ve: Negative control). [Click here to view] |
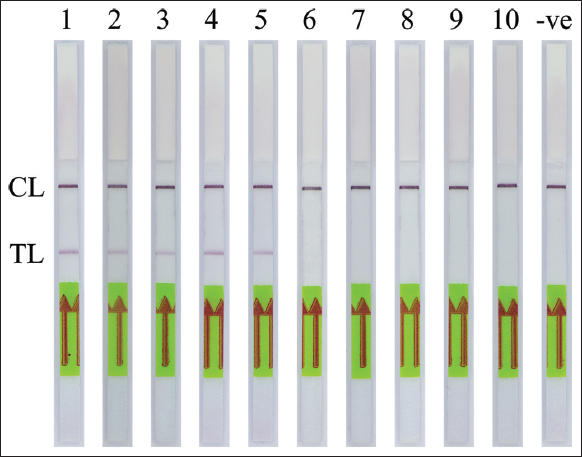 | Figure 9: The specificity of polymerase chain reaction-nucleic acid lateral flow immunoassay assay for detection of the human SRY gene from saliva samples. CL: Control line, TL: Test line (1–5: Male saliva samples, 6–10: Female saliva samples and -ve: Negative control). [Click here to view] |
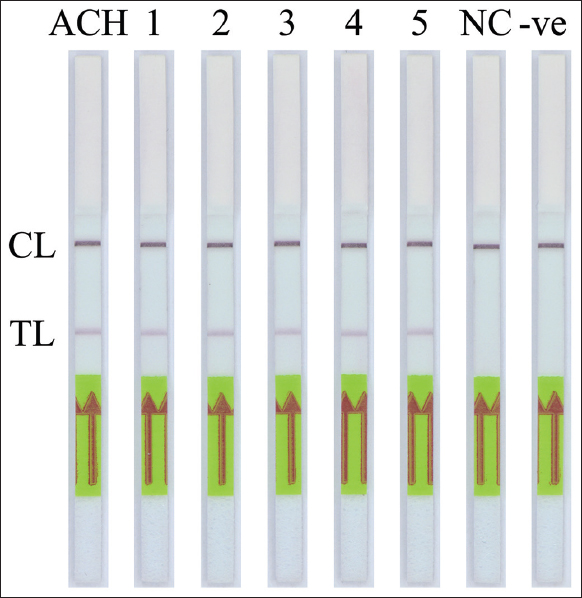 | Figure 10: The specificity of AS-polymerase chain reaction-nucleic acid lateral flow immunoassay assay for detection of the heterozygous G1138A mutation in the FGFR3 gene. CL: Control line, TL: Test line. (ACH: Positive control for heterozygous G1138A mutation, 1–5: Patients with ACH (heterozygous G1138A mutation), NC: Normal individual; -ve: Negative control). [Click here to view] |
4. DISCUSSION
This research investigated the development of PCR-NALFIA assay based on human SRY gene for sex determination and AS-PCR-NALFIA assay for detecting heterozygous G1138A mutation in the FGFR3 gene for the diagnosis of achondroplasia (ACH). In this research, the PCR-NALFIA assay was designed to detect male DNA and assessed for its property to identify human SRY gene in human blood, semen, and saliva samples. Moreover, AS-PCR-NALFIA assay for detecting heterozygous G1138A mutation in the FGFR3 gene was also successfully identified in the human DNA sample. The NALFIA system combines both PCR amplification and universal LFD format. The result can be interpreted on the LFD within 5 min with the naked eye. Compared to conventional amplicon detection methods, the LFD system does not need special tools, gel electrophoresis equipment, or post-amplification processing. As a result, the NALFIA system can probably be considered a promising new molecular tool for DNA detection in a moderately well-equipped laboratory with standard thermal cycler apparatus and refrigeration.
For PCR-NALFIA assay used to detect human SRY gene, specific primers for SRY gene, including SRYF and SRYR labeled with biotin and FAM at the 5′ ends, were used for PCR-NALFIA. Male and female DNA samples were used as a template to amplify human SRY gene. The specific double-labeled PCR amplicons achieved with the SRYF/SRYR primers were also detected using the NALFIA. The LFD provided positive results when using male DNA. In contrast, the LFD displayed negative results when using female DNA as a template for PCR reactions. Although the specific double-labeled amplicon was invisible in GE method, a positive response was accomplished using the specific double-labeled amplicon as an analyte in NALFIA system. Then, the purified double-labeled amplicon was quantified by using Qubit™ 4 fluorometer (Invitrogen by Thermo Fisher Scientific, USA), QFX fluorometer (DeNovix, USA), and DS-11 spectrophotometer (DeNovix, USA). It revealed that the concentration of the purified double-labeled amplicon was below the limit. This solution indicated that the sensitivity of the PCR-NALFIA assay was higher than agarose GE for the visualization of double-labeled PCR amplicons. However, PCR products generated from regular primers showed the band only at the control line on the LFD, although 220 bp fragments of SRY gene are present with male DNA. For PCR-NALFIA assay used to detect G1138A mutation in FGFR3 gene, specific primers, including ACHF and ACHR labeled with biotin and FAM at the 5′ end, were used for AS-PCR-NALFIA. DNA samples from patients with ACH (heterozygous G1138A mutation) and normal individuals were used as a template to amplify FGFR3 gene. The double-labeled ASPCR amplicon was invisible when the GE method was performed. However, a positive response was accomplished using the double-labeled ASPCR product as an analyte in the NALFIA system, providing the same result as the PCR-NALFIA. However, ASPCR products generated from regular primers showed the band only at the control line on the LFD, although 122 bp fragments of the FGFR3 gene are present with individual heterozygous G1138A mutation. Moreover, the sequencing analysis of the specific purified PCR/ASPCR products from PCR/ASPCR reactions from both regular primers confirmed the expected size and sequence.
These outcomes confirm the high detection sensitivity of the PCR- and AS-PCR-NALFIA systems developed in this study. When the PCR amplification utilizes a DNA target and specific primers labeled with biotin and FAM at the 5′ end, the sensitivity level does not decrease. This investigation supports that the sensitivity of both the PCR- and AS-PCR-NALFIA assays is higher than that of PCR-GE, which is in accordance with other works [30,31]. As previously reported, PCR-NALFIA assay sensitivity was over 1000 times greater than PCR-GE [31].
The detection limit of both PCR- and AS-PCR-NALFIA assays was assessed by using different levels of concentration of the DNA template. The detection limit of both the PCR- and AS-PCR-NALFIA systems developed in this study was 1 ng, where a faint test line was still visible on the LFD in the NALFIA system. PCR-GE was also performed simultaneously, but the double-labeled PCR product was invisible on the agarose gel. These outcomes confirmed that the employed universal LFD had a higher sensitivity. Regarding the limits of detection, PCR-NALFIA was superior [32]. According to a previous study, the ribosomal DNA intergenic spacer sequences used in the NALFIA assay to detect the fungus Macrophomina phaseolina in agricultural soil samples had a detection limit of 17.3 fg [31]. Moreover, as previously reported, the detection limit for the verification of sheep-specific PCR products was 0.01 pg based on the mitochondrial gene sequences of sheep [30]. It could be stated that the higher copy number of both areas increases the sensitivity of PCR-based approaches. Therefore, this may be the reason why differs from our results because both target genes used in PCR- and AS-PCR-NALFIA assays have a lower number of copies. From our results, it could be determined that the universal LFD system achieves a high detection sensitivity as a sensitive molecular technology. However, there is a chance that aerosols may have contaminated the sample, thus producing false-positive results [30]. This issue is considered indispensable in PCR-NALFIA with the universal LFD because the method is even more sensitive than PCR-GE. However, the LFD still has some benefits besides its greater sensitivity compared to GE detection, such as the quick test that could be completed within 5 min. It is considerably easy to carry out and does not require expensive equipment or trained personnel. Moreover, the interpretation method is simple. PCR-NALFIA is a specific and sensitive method that requires only about 1.5–2 h from DNA extraction to detection of the target genes [32]. Due to the necessity for a quick and sensitive detection technique that utilizes a POC testing strategy, the utilization of the LFD system for organism detection is gradually becoming more common in several scientific areas [31].
5. CONCLUSION
From the results found in this study, it could be stated that the developed PCR- and AS-PCR-NALFIA assays in the study were suitable for identifying target gene in DNA samples. The detection limit of both PCR- and AS-PCR-NALFIA assays was determined to be 1 ng for template DNA. The PCR-NALFIA assay was specific for human SRY gene detection, and cross-contaminations were not detected with any non-target genes. In addition, AS-PCR-NALFIA assay was also specific for detecting heterozygous G1138A mutation in the FGFR3 gene to diagnose achondroplasia (ACH). This NALFIA system can be quickly altered and used with different target genes. Therefore, it might be practical for gene detection in various contexts as a simple-to-use system. Furthermore, this procedure could become a practical alternative for advancing rapid diagnostic kits for the target gene.
6. ACKNOWLEDGMENTS
The authors would like to thank the staff from the Srinagarind hospital and the Department of Biology, Faculty of Science, Khon Kaen University, Khon Kaen, Thailand for sample collection and laboratory equipment providing.
7. AUTHORS’ CONTRIBUTIONS
All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; agreed to submit to the current journal; gave final approval of the version to be published; and agreed to be accountable for all aspects of the work. All the authors are eligible to be an author as per the International Committee of Medical Journal Editors (ICMJE) requirements/guidelines.
8. FUNDING
The project and manuscript preparation were financially supported by the Science Achievement Scholarship of Thailand (SAST) and Khon Kaen University.
9. CONFLICTS OF INTEREST
The authors report no financial or any other conflicts of interest in this work.
10. ETHICAL APPROVALS
This study was approved by the Khon Kaen University Ethics Committee for Human Research (approval no. HE631576).
11. DATA AVAILABILITY
All data generated and analyzed are included within this research article.
12. PUBLISHER’S NOTE
This journal remains neutral with regard to jurisdictional claims in published institutional affiliation.
REFERENCES
1. Posthuma-Trumpie GA, Korf J, van Amerongen A. Lateral flow (immuno) assay:Its strengths, weaknesses, opportunities and threats. A literature survey. Anal Bioanal Chem 2009;393:569-82. [CrossRef]
2. Ngom B, Guo Y, Wang X, Bi D. Development and application of lateral flow test strip technology for detection of infectious agents and chemical contaminants:A review. Anal Bioanal Chem 2010;397:1113-35. [CrossRef]
3. Bahad?r EB, Sezgintürk MK. Lateral flow assays:Principles, designs and labels. TrAC Trends Anal Chem 2016;82:286-306. [CrossRef]
4. Mens PF, Moers A, de Bes LM, Flint J, Sak JR, Keereecharoen L, et al. Development, validation and evaluation of a rapid PCR-nucleic acid lateral flow immuno-assay for the detection of Plasmodium and the differentiation between Plasmodium falciparum and Plasmodium vivax. Malar J 2012;11:279. [CrossRef]
5. Roth JM, de Bes L, Sawa P, Omweri G, Osoti V, Oberheitmann B, et al. Plasmodium detection and differentiation by direct-on-blood PCR nucleic acid lateral flow immunoassay. J Mol Diagn 2018;20:78-86. [CrossRef]
6. Rule GS, Montagna RA, Durst RA. Rapid method for visual identification of specific DNA sequences based on DNA-tagged liposomes. Clin Chem 1996;42:1206-9. [CrossRef]
7. Wurmb-Schwark NV, Bosinski H, Ritz-Timme S. What do the X and Y chromosomes tell us about sex and gender in forensic case analysis?J Forensic Leg Med 2007;14:27-30. [CrossRef]
8. Masuyama K, Shojo H, Nakanishi H, Inokuchi S, Adachi N. Sex determination from fragmented and degenerated DNA by amplified product-length polymorphism bidirectional SNP analysis of amelogenin and SRY genes. PLoS One 2017;12:e0169348. [CrossRef]
9. Butler E, Li R. Genetic markers for sex identification in forensic DNA analysis. J Forensic Investig 2014;2:10.
10. Hrovatin K, Kunej T. Genetic sex determination assays in 53 mammalian species:Literature analysis and guidelines for reporting standardization. Ecol Evol 2018;8:1009-18. [CrossRef]
11. Maulani C, Auerkari EI. Molecular analysis for sex determination in forensic dentistry:A systematic review. Egypt J Forensic Sci 2020;10:36. [CrossRef]
12. Drobni?K. A new primer set in a SRY gene for sex identification. Int Congr Ser 2006;1288:268-70. [CrossRef]
13. Kholief M, El Shanawany S, Gomaa R. Sex determination from dental pulp DNA among Egyptians. Egypt J Forensic Sci 2017;7:29. [CrossRef]
14. Tozzo P, Giuliodori A, Corato S, Ponzano E, Rodriguez D, Caenazzo L. Deletion of amelogenin Y-locus in forensics:Literature revision and description of a novel method for sex confirmation. J Forensic Leg Med 2013;20:387-91. [CrossRef]
15. Nogami H, Tsutsumi H, Komuro T, Mukoyama R. Rapid and simple sex determination method from dental pulp by loop-mediated isothermal amplification. Forensic Sci Int Genet 2008;2:349-53. [CrossRef]
16. Kanchanaphum P. Time course of detection of human male DNA from stained blood sample on various surfaces by loop mediated isothermal amplification and polymerase chain reaction. Biomed Res Int 2018;2018:2981862. [CrossRef]
17. Cadenas AM, Regueiro M, Gayden T, Singh N, Zhivotovsky LA, Underhill PA, et al. Male amelogenin dropouts:Phylogenetic context, origins and implications. Forensic Sci Int 2007;166:155-63. [CrossRef]
18. Kailasa SK, Mehta VN, Koduru JR, Basu H, Singhal RK, Murthy ZV, et al. An overview of molecular biology and nanotechnology based analytical methods for the detection of SARS-CoV-2:Promising bio-tools for the rapid diagnosis of COVID-19. Analyst 2021;146:1489-513. [CrossRef]
19. Surti PV, Kim MW, Phan LM, Kailasa SK, Mungray AK, Park JP, et al. Progress on dot-blot assay as a promising analytical tool:Detection from molecules to cells. TrAC Trends Anal Chem 2022;157:116736. [CrossRef]
20. Pauli RM. Achondroplasia:A comprehensive clinical review. Orphanet J Rare Dis 2019;14:1. [CrossRef]
21. Richette P, Bardin T, Stheneur C. Achondroplasia:From genotype to phenotype. Joint Bone Spine 2008;75:125-30. [CrossRef]
22. Xue Y, Sun A, Mekikian PB, Martin J, Rimoin DL, Lachman RS, et al. FGFR3 mutation frequency in 324 cases from the International Skeletal Dysplasia Registry. Mol Genet Genomic Med 2014;2:497-503. [CrossRef]
23. Bellus GA, Hefferon TW, de Luna O, Hecht JT, Horton WA, MachadoM. Achondroplasia is defined by recurrent G380R mutations of FGFR3. Am J Hum Genet 1995;56:368-73.
24. Patil SJ, Banerjee M, Phadke SR, Mittal B. Mutation analysis in Indian children with achondroplasia-utility of molecular diagnosis. Indian J Pediatr 2009;76:147-9. [CrossRef]
25. Su YN, Lee CN, Chien SC, Hung CC, Chien YH, Chen CA. Rapid detection of FGFR3 gene mutation in achondroplasia by DHPLC system-coupling heteroduplex and fluorescence-enhanced primer-extension analysis. J Hum Genet 2004;49:399-403. [CrossRef]
26. Schrijver I, Lay MJ, Zehnder JL. Rapid combined genotyping assay for four achondroplasia and hypochondroplasia mutations by real-time PCR with multiple detection probes. Genet Test 2004;8:185-9. [CrossRef]
27. He X, Xie F, Ren ZR. Rapid detection of G1138A and G1138C mutations of the FGFR3 gene in patients with achondroplasia using high-resolution melting analysis. Genet Test Mol Biomarkers 2012;16:297-301. [CrossRef]
28. Imtawil K, Muisook K, Namwat N, Loilome W, Tangrassameeprasert R, Wichajarn K. Detection of the G1138A mutation in the FGFR3 gene for the diagnosis of achondroplasia by allele-specific polymerase chain reaction. J Med Assoc Thai 2019;102:118-21.
29. Gomes ME, Kanazawa TY, Riba FR, Pereira NG, Zuma MC, Rabelo NC, et al. Novel and recurrent mutations in the FGFR3gene and double heterozygosity cases in a cohort of Brazilian patients with skeletal dysplasia. Mol Syndromol 2018;9:92-9. [CrossRef]
30. Yin R, Sun Y, Yu S, Wang Y, Zhang M, Xu Y, et al. A validated strip-based lateral flow assay for the confirmation of sheep-specific PCR products for the authentication of meat. Food Control 2016;60:146-50. [CrossRef]
31. Pecchia S, Da Lio D. Development of a rapid PCR-nucleic acid lateral flow immunoassay (PCR-NALFIA) based on rDNA IGS sequence analysis for the detection of Macrophomina phaseolina in soil. J Microbiol Methods 2018;151:118-28. [CrossRef]
32. Seidel C, Peters S, Eschbach E, Feßler AT, Oberheitmann B, Schwarz S. Development of a nucleic acid lateral flow immunoassay (NALFIA) for reliable, simple and rapid detection of the methicillin resistance genes mecA and mecC. Vet Microbiol 2017;200:101-6. [CrossRef]